Это очень редкое заболевание, его находят у одного из 10 миллионов младенцев. Названо оно по именам бразильского эндокринолога У. Бернанденелли и норвежского педиатра М. Сейпа, которые его и описали. Другие его названия генерализованная липодистрофия, липоатрофический диабет, акромегалоидный гигантизм.
Эта врожденная болезнь характеризуется отсутствием жировой ткани. Существует два типа синдрома Берардинелли–Сейпа: в первом случае жировая прослойка присутствует в суставах, на ладонях и подошвах, выполняя амортизирующую функцию, во втором – она полностью отсутствует.
Причиной заболевания является мутация генов.
Проявления синдрома Берардинелли–Сейпа
Дети, рождающиеся с этим недугом, кроме отсутствия подкожной жировой клетчатки, обладают и другими признаками. Это очень быстрый рост, усиленный аппетит, увеличенная нижняя челюсть, крупные стопы и ладони, усиленный рост терминальных (черных и жестких) волос на лице, шее, руках и ногах. Тем не менее при отсутствии подкожного жира больные не выглядят дистрофиками, так как мышцы у них развиты достаточно хорошо.
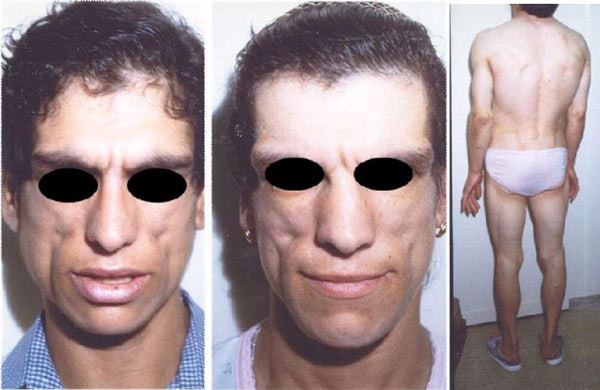
Осложнения синдрома Берардинелли–Сейпа
Заболевание вызывает нарушения и патологии в различных органах и системах. Так, у больных часто находят:
- увеличенную в размерах печень;
- увеличенную селезенку;
- пупочную грыжу;
- повышенное содержание липидов в крови;
- уже в юношеском возрасте сахарный диабет 2-го типа;
- у девушек – увеличенный клитор, поликистоз яичников;
- при синдроме второго типа – легкую умственную отсталость;
- в области шеи черный акантоз – дистрофическое заболевание кожи, характеризующееся изменением ее цвета на серый или черный и появлением мелких папиллом;
- кисты костей;
- кардиомиапатию – увеличение размера сердца, нарушения ритма, сердечную недостаточность.
Лечение синдрома Берардинелли–Сейпа
Вылечить само заболевание невозможно. Помощь страдающим наследственной липодистрофией заключается в лечении многочисленных осложнений, в первую очередь – сахарного диабета, печеночной и сердечной недостаточности.