
В настоящее время изучено большинство молекулярных и клеточных механизмов, ответственных за сердечно-сосудистые эффекты гормонов щитовидной железы. Для понимания изменений сердечно-сосудистой системы под действием её гормонов, важно знать механизмы действия тироидных гормонов на кардиомиоциты и гладкомышечные клетки сосудов.
Тироксин (Т4) и трийодтиронин (Т3) синтезируются щитовидной железой под воздействием тиреотропного гормона (ТТГ). Щитовидная железа преимущественно секретирует Т4 (85%), который превращается в Т3 под действием 5-монодейодиназы в печени, почках, гипофизе, коре надпочечников и скелетной мускулатуре [1]. Нет доказательств того, что переход тироксина в трийодтиронин происходит в кардиомиоците, предполагают, что состояние сердца зависит от уровня сывороточного Т3, и мембрана клеток содержит определенные транспортные белки для него [2]. В кардиомиоците Т3 входит в ядро, где связывается с ядерным рецептором (TRs), и этот комплекс, в свою очередь, связывается с элементами, отвечающими за гормональный ответ (TREs) на промоутерных зонах регуляторных генов. TRs относится к суперсемейству рецепторов к стероидным гормонам, но, в отличие от них, TRs может связываться с TREs как в присутствии, так и в отсутствие лиганда (Т3). В присутствии Т3 TRs индуцирует транскрипцию генов, а в отсутствие Т3 подавляет ее [3]. Негативно регулируемые сердечные гены, такие как гены бета-тяжелые цепей миозина и фосфоламбана, активируются в отсутствие Т3 и подавляются в его присутствии [4,5].
Эффект тиреоидных гормонов на кардиомиоциты тесно связан с регуляцией экспрессии ключевых структурных и регуляторных генов. Гены тяжелых цепей миозина кодируют 2 изоформы сократительного белка толстого филамента кардиомиоцита. Ca АТФаза саркоплазматического ретикулума и ее ингибитор фосфоламбан регулируют внутриклеточную циркуляцию кальция. Вместе они в значительной степени ответственны за сократительную функцию и диастолическое расслабление сердца. Активация бета-адренергических рецепторов и активность Na/K-АТФазы также регулируются Т3 [6].
Прямые эффекты гормонов щитовидной железы на сердце
Тиреоидные гормоны являются важными регуляторами экспрессии сердечных генов, и многие сердечные проявления дисфункции щитовидной железы связаны с изменениями экспрессии Т3-зависимых генов. И у человека, и у животных тиреотоксикоз приводит к гипертрофии миокарда. Эти изменения, в первую очередь, являются результатом увеличенной работы сердца и повышением гемодинамической нагрузки [7].
Трийодтиронин-представляющие гены, кодирующие структурные и регулирующие белки в сердце, перечислены в таблице. Два миозина тяжелых цепей (альфа- и бета-) являются мышечными белками, которые составляют основу мышечного аппарата кардиомиоцитов. Альфа-тяжелые цепи миозина относятся к быстрому миозину с повышенной АТФазной активностью, а бета-тяжелые— к медленному миозину. У животных Т3 активирует транскрипцию генов миозина тяжелых цепей альфа-, в то время как транскрипция генов миозина тяжелых цепей бета- подавляется. У людей преобладает миозин тяжелых цепей бета-, и хотя сократительная функция сердца заметно меняется при патологии щитовидной железы, изменения соотношения изоформ миозина тяжелых цепей недостаточно, чтобы вызвать функциональные сдвиги [8, 9].
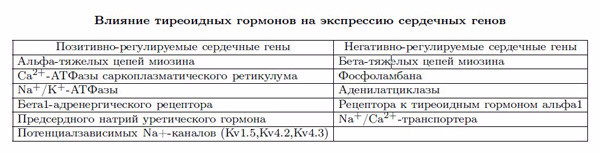
Экспрессия генов Са-АТФазы саркоплазматического ретикулума и ее ингибитора фосфоламбана находится под контролем Т3. Вход кальция и его перераспределение в саркоплазматическом ретикулуме являются критическими детерминантами систоли- ческой сократительной функции и диастолического расслабления [10, 11]. Активный транспорт кальция Са-АТФазой в пластинку саркоплазматического ретикулума регулируется фосфоламбаном, чья деятельность, в свою очередь, зависит от уровня фосфорилирования [11]. Таким образом, изменения относительного количества этих белков и степень фосфорилирования фосфламбана при патологии щитовидной железы изменяют диастолическую функцию. У трансгенных мышей, испытывающих недостаток фосфоламбана, было выявлено увеличение сердечной сократимости, и лечение тироидными гормонами не давало дополнительного инотропного эффекта [12]. Этот результат подтверждает роль фосфоламбана в индуцированных тироидными гормонами изменениях сократимости и объясняет увеличение диастолической функции у пациентов с тиреотоксикозом [7].
Учащение сердечных сокращений, расширение пульсового давления и увеличение сердечной продукции у пациентов с тиреотоксикозом связаны со степенью усиления адренергической активации, несмотря на нормальную или сниженную концентрацию катехоламинов [13]. Исследования различных компонентов адренергического рецепторного комплекса в плазматической мембране выявили при нарушении функции щитовидной железы изменения работы бета-адренорецепторов, гуаниннуклеотид-регулирующих белков, аденилатциклазы типа V и VI. Активность некоторых мембранных ионных транспортеров, таких как Na/K-ATФaза, Na/Ca-транспортер и К-каналы, также регулируется уровнем содержания гормонов щитовидной железы, координируя, таким образом, электрохимические и механические ответы миокарда [14, 15].
Гормоны щитовидной железы оказывают также и внеядерное действие на кардиомиоциты. В отличие от эффектов, связанных с воздействием на ядерный рецептор, появление которых занимает от 30 минут до 2 часов, изменения в работе ионных каналов под действием Т3 начинаются в течение нескольких минут. Т3 увеличивает активность сердечной Na/K-АТФазы. Этот фермент вытесняет натрий из клетки в обмен на внеклеточный калий. Кроме того, существуют немногочисленные данные о влиянии Т3 на трансмембранный транспорт глюкозы и аминокислот. В короткие сроки трийодтиронин изменяет особенности работы натриевых, калиевых и кальциевых каналов в сердце и через изменения во внутриклеточных уровнях содержания кальция и калия может усиливать инотропный и хронотропный эффекты [16]. Таким образом, и транскрипционный, и нетранскрипционный эффекты тироидных гормонов могут осуществляться в совокупности, модулируя функцию миокарда и сосудистой системы в физиологических и патофизиологических условиях [7].
В исследованиях Nathan A. et al (1983) были получены данные о том, что при диффузном токсическом зобе (болезни Грейвса) поражение сердца и развитие миокардиодистрофии связаны не только с действием тироидных гормонов, но и с присутствием особых аутоантител к кардиомиоцитам, которые оказывают стимулирующее действие на миокард и способствуют входному току кальция в его клетки [17].
Влияние гормонов щитовидной железы нагемо динамику
Эффект действия тироидных гормонов на сердце и периферические сосуды включает уменьшение общего периферического сосудистого сопротивления (ОПСС), увеличение частоты сердечных сокращений в покое, левожелудочковой сократимости и объема крови. Тироидные гормоны, действуя непосредственно на гладкомышечные клетки, вызывают расширение сосудов гладкомышечного типа и снижение среднего артериального давления. В ответ на это в почках происходит активация ренин-ангиотензин-альдостероновой системы и увеличение почечной абсорбции натрия. Также Т3 увеличивает синтез эритропоэтина, что приводит к увеличению эритроцитарной массы. Вместе эти изменения приводят к увеличению объема циркулирующей крови (ОЦК) и преднагрузки. При тиреотоксикозе эти эффекты увеличивают сердечный выброс на 50–300% по сравнению со здоровыми людьми. При гипотиреозе эти эффекты диаметрально противоположны, и сердечный выброс может уменьшиться на 30–50% [6].
Сосудорасширяющее действие тироидных гормонов является результатом ответной реакции эндотелиальных клеток сосудов, выделяющих оксид азота, обладающий расслабляющим влиянием на гладкомышечные клетки сосудов. При гипотиреозе происходит увеличение ОПСС. Нарушение эндотелий-зависимой вазодилятации в результате снижения синтеза оксида азота хорошо демонстрируется при субклиническом гипотиреозе [18]. При тиреотоксикозе ОПСС снижается, а ОЦК и перфузия в периферических тканях возрастают. Наблюдение повышенной васкуляризации, сопровождающей тиреотоксикоз, дает основание предполагать, что Т3 может увеличивать плотность капилляров и через усиление ангиогенеза [6]. При тиреотоксикозе также обнаружено повышение уровня адреномедуллина, являющегося мощным вазодилятатором [19].
Ренин-ангиотензин-альдостероновая система играет важную роль в регуляции кровяного давления. Юкстагломерулярный аппарат почек является объемо- и давление-зависимым, и в ответ на снижение среднего артериального давления происходит активация ренин-ангиотензин-альдостероновой системы, и секреция ренина увеличивается. Далее следует каскад событий, включающий в себя повышение уровня ангиотензина I и II, ангиотензин-превращающего фермента и альдостерона. Поэтому, в то время как тироидные гормоны снижают ОПСС и постнагрузку, усиление секреции ренина и альдостерона приводит к увеличению ОЦК и преднагрузки, что вносит свой вклад в характерное увеличение сердечного выброса [20, 21].
Напротив, при гипотиреозе, несмотря на повышение тонуса и сопротивление кровотоку в периферических сосудах, среднее гемодинамическое давление крови не изменяется, систолическое давление в результате пониженной насосной функции сердца может быть сниженным, а диастолическое— повышенным и, как результат, пульсовое давление понижается. Увеличение диастолического давления сопровождается низким уровнем ренина и является соль-чувствительной формой гипертензии [22]. У пациентов с тиреотоксикозом концентрация эритропоэтина повышена, хотя гематокрит и уровень гемоглобина остаются нормальными, что связано с сопутствующим увеличением ОЦК. А при гипотиреозе уровень эритропоэтина снижается, что может объяснить нормохромную анемию, выявляемую у 35% таких пациентов [6].
Эффект действия гормонов щитовидной железы нарегуляцию артериального давления
Популяционные исследования указывают на изменения артериального давления при любой дисфункции щитовидной железы [23]. Asvold et al. сообщают о линейной корреляции между уровнем ТТГ и уровнем систолического и диастолического давления [24], однако другие исследования не подтверждают эти данные. Тироидные гормоны повышают основной обмен, что приводит к изменениям сердечного выброса, ОПСС и артериального давления [23].
Долгое время считалось, что тиреотоксикоз сопровождается значительным повышением систолического давления и значимым снижением диастолического. Однако в 1960-х годах Л. М. Гольбер и В. И. Кандрор показали, что при измерении артериального давления прямым методом, т. е. с помощью электроманометра в условиях катетеризированной сонной артерии, отмечается повышение как систолического, так и диастолического давления. Причина расхождения кроется, по-видимому, в различии используемых методов измерения артериального давления. Еще в 1911 г. М. В. Яновский описал феномен «бесконечного тона», т. е. сохранения коротковских тонов даже при падении давления в компрессионной манжете до нуля. Исследования Г. И. Косицкого (1959) обнаружили «бесконечный тон» при реальном диастолическом давлении равном 60 ммрт. ст. Все это позволяет сделать заключение, что мнение о падении диастолического давления при тиреотоксикозе является ошибочным и связано с использованием непрямого метода измерения давления [25].
Тиреотоксикоз признан одной из причин вторичной изолированной систолической гипертензии, которая является самой частой формой гипертензии [26]. Лечение тиреотоксикоза и использование бета-блокаторов уменьшают частоту сердечных сокращений и устраняют эти изменения.
При гипотиреозе эндотелиальная дисфункция и нарушения расслабления гладко-мышечных сосудов ведут к увеличению ОПСС. Эти эффекты приводят к диастолической гипертензии у 30% пациентов, а заместительная гормональная терапия восстанавливает эндотелий-зависимую релаксацию и в большинстве случаев нормализует артериальное давление [6].
Влияние гормонов щитовидной железы налипидный обмен
Хорошо известно, что гипотиреоз сопровождается повышением уровня липидов. Явный гипотиреоз сопровождается гиперхолестеринемией с увеличением уровня липопротеидов низкой плотности (ЛПНП) и аполипопротеинов В. Распространенность явного гипотиреоза у пациентов с гиперхолестеринемией составляет от 1,3 до 2,8%, а 90% пациентов с гипотиреозом имеют гиперхолестеринемию. Даже ранние стадии гипотиреоза сопровождаются изменением уровня липидов. Ряд исследований указывает на увеличение уровня ЛПНП при субклиническом гипотиреозе, однако в других исследованиях выявляется повышение уровня общего холестерина без изменения ЛПНП. Механизм гиперхолестеринемии при гипотиреозе связан со снижением содержания рецепторов ЛП-НП в печени и вследствие этого— уменьшением печеночной экскреции холестерина и, далее, повышением уровня ЛПНП и липопротеидов очень низкой плотности (ЛПОНП) [27]. Повышение уровня липидов при субклиническом и явном гипотиреозе ассоциировано с повышением кардиоваскулярного риска. Лечение тироидными гормонами и восстановление эутиреоидного состояния полностью нивелирует этот риск [28].
Амиодарон и функция щитовидной железы
Амиодарон— богатый йодом антиаритмический препарат с доказанной эффективностью в лечении пациентов с желудочковыми и предсердными аритмиями. Приблизительно 37% массы амиодарона приходится на органический йод, из них 10%— в форме дийодида. Стандартный прием амиодарона (0,2 г/сут) приводит к ежедневному потреблению органического йода в дозе 0,075 г. У пациентов, принимающих амиодарон, уровень неорганического йода в моче и плазме повышен в 40 раз. Амиодарон блокирует превращение тироксина в трийодтиронин в большинстве, если не во всех, тканей. Имея высокую концентрацию йода, он может также блокировать синтез и секрецию тиреоидных гормонов. У пациентов с нормальной тиреоидной функцией, получающих амиодарон, сывороточная концентрация трийодтиронина снижается на 20–25% и остается низкой. Концентрация тироксина и ТТГ увеличивается выше нормального диапазона, но иногда может несколько снижаться [29]. У 5–25% пациентов развивается амиодарониндуцированный гипотиреоз. Установлено, что большинство пациентов этой группы уже имели аутоиммунный тиреоидит, который нарушает нормальное восстановление щитовидной железы при действии йода [30]. Резкое изменение йодного баланса при лечении амиодароном может повлиять на альтернативный процессинг и условия иммунологической презентации тироглобулина. В связи с этим важно отметить, что имеются исследования, свидетельствующие об увеличении титра антител к тиреоидной пероксидазе и тироглобулину на фоне длительного приема амиодарона [31, 32].
Тиреотоксикоз при лечении амиодароном развивается в 2–10% случаев. Поэтому, учитывая эти данные, в первый год лечения амиодароном контроль ТТГ должен осуществляться каждые 2 месяца и реже— в последующие годы лечения [29, 30].
Существует две формы амиодарон-индуцированного тиреотоксикоза (1 и 2 тип). Тип 1 тиреотоксикоза обычно развивается у пациентов с узловыми зобами, которые проживают в регионе легкого йододефицита, и связан с высокой концентрацией йода в амиодароне. Тиреотоксикоз типа 2 развивается у пациентов с нормальной щитовидной железой в результате развития деструктивного тиреоидита. Окончательно механизм этого процесса не известен, однако, возможно, это происходит на фоне развития аутоиммунных процессов и под воздействием ИЛ-6 [34]. Пациенты с тиреотоксикозом типа 1 имеют нормальные или даже высокие 24-часовые кривые захвата йода щитовидной железой, и поэтому они могут лечиться радиойодом. Альтернативно они могут получать лечение антитиреоидными препаратами [35]. Пациенты со 2 типом тиреотоксикоза имеют нормальную или несколько увеличенную щитовидную железу и низкий захват йода щитовидной железой. Наиболее эффективна терапия для этих пациентов— преднизолон в дозе 30–40 мг в сутки [34]. Обычно улучшение наступает в течение нескольких недель, после чего доза может постепенно уменьшаться. Лечение антитиреоидными препаратами чаще всего неэффективно. Однако бета-адреноблокаторы могут быть полезны, особенно у пациентов с тахиаритмиями или сниженной толерантностью к нагрузке из-за мышечной слабости [35].
Заключение
Гормоны щитовидной железы оказывают прямое и опосредованное влияние на сердечно-сосудистую систему. Прямые эффекты связаны с воздействием на транскрипцию генов и внеядерным действием на работу натриевых, кальциевых и калиевых каналов. Опосредованные эффекты, такие как изменение ОПСС, артериального давления, развиваются в результате активации систем, ответственных за поддержание сердечно-сосудистого гомеостаза, в частности, ренин-альдостерон-ангиотензиновой системы. Под действием этих изменений у пациентов с патологией щитовидной железы, особенно с тиреотоксикозом, признаки и симптомы, указывающие на изменения сердечно-сосудистой системы, выходят на первое место. Однако данные изменения являются обратимыми, и нормализация функции щитовидной железы приводит к восстановлению основных параметров.
Литература
- Левина Л. И. Сердце при эндокринных заболеваниях. Л.: Медицина, 1989.
- Кандрор В.И. Молекуляpно-генетические аспекты тиреоидной патологии // Пробл. эндокринологии. 2001. Т. 47, № 5. С. 3–10.
- Lazar M.A., Chin W.W. Nuclear thyroid hormone receptors // J. Clin. Invest. 1990. Vol. 86. Р. 1777–1782.
- Danzi S, Dubon P, Klein I. Effect of serum T3 on the regulation of cardiac gene expression: role of histone acetylation // Am. J. Physiol. Heart Circ. Physiol. 2005. Vol. 289. Р. 1506–1511.
- Hu X., Lazar M. A. Transcriptional repression by nuclear hormone receptors // Trends Endocrinol Metab. 2000. Vol. 11. Р. 6–10.
- Klein I, Danzi S. Thyroid disease and the heart // Circulation 2007. Vol. 116. Р. 1725–1735.
- Klein I., Ojamaa K. Thyroid hormone and the cardiovascular system // N. Engl. J. Med. 2001. Vol. 344. Р. 501–509.
- Ojamaa K., Klemperer J. D., MacGilvray S. S., Klein I., Samarel A. Thyroid hormone and hemodynamic regulation of beta-myosin heavy chain promoter in the heart // Endocrinology 1996. Vol. 137. Р. 802–808.
- Morkin E. Regulation of myosin heavy chain genes in the heart // Circulation. 1993. Vol. 87. Р. 1451–1460.
- Dillmann W.H. Biochemical basis of thyroid hormone action in the heart // Am. J. Med. 1990. Vol. 88. Р. 626–630.
Источник: Карась А.С., Обрезан А.Г. Влияние гормонов щитовидной железы на сердце: молекулярные, клеточные, тканевые и органные аспекты (обзор литературы) / // Вестник Санкт-Петербургского Университета, серия 11, медицина. – СПб., 2009. – Вып. 4. – С. 28–35.
