
Современные достижения медицины в значительной степени отражают мощное и поступательное развитие кардиологической науки и практики. Скорость обмена информации в такой области медицинских знаний как «кардиология» очень велика. Открытие новых возбудителей сердечно-сосудистых заболеваний, описание новых путей патогенеза, внедрение современных инструментальных и лабораторных диагностических критериев существенно продвигают эту отрасль медицины. Наряду с данными прогрессивными изменениями врачу-кардиологу приходится постоянно встречаться с дилеммой устаревания одних терминов и одновременного внедрения иных. Процесс терминологической замены зачастую принимает характер императивного перенесения отдельных зарубежных переводных понятий в отечественную врачебную практику. В этом инновационном процессе, к сожалению, забывается отечественный приоритет в изучении тех или иных заболеваний, в исторически традиционном придании им нозологической самостоятельности. В настоящей статье пойдет речь о терминологических проблемах в одном из разделов некоронарогенных заболеваний миокарда.
Термин «кардиомиопатии» (КМП) впервые был предложен W. Brigden в 1957 г. для обозначения первичных поражений миокарда неизвестной этиологии, вызывающих нарушение функции сердца и не являющихся следствием заболеваний коронарных артерий, клапанного аппарата, перикарда, системной или легочной гипертензии, а также некоторых редких вариантов поражения проводящей системы сердца. В понимании W. Brigden КМП имела самостоятельность и не была ассоциирована с известными причинными факторами, в те годы генетических причин КМП также не описывали [1].
J. Goodwin в 1989 г. уточнил приведенное положение о кардиомиопатиях как о самостоятельной, идиопатической, проградиентно текущей необратимой нозологии, имеющей четкое наследственное сцепление и первичный характер поражения сердечной мышцы. В определении J. Goodwin содержатся важнейшие критерии КМП и прежде всего отсутствие зависимости от какой-либо четко определяемой (известной) причины [2].
Согласно классификации J. Goodwin, различают 3 группы КМП:
- Дилатационная КМП (ДКМП) — характеризуется значительной дилатацией камер сердца, систолодиастолической дисфункцией миокарда и отсутствием выраженной гипертрофии сердечной мышцы. Гемодинамика при ДКМП страдает вследствие нарушения систолической функции (снижения фракции выброса левого желудочка (ЛЖ)).
- Гипертрофическая КМП (ГКМП) — отличается значительной, чаще асимметричной, гипертрофией миокарда левого и/или правого желудочков, отчетливым преобладанием диастолической дисфункции миокарда ЛЖ (ДДЛЖ) и отсутствием дилатации полостей сердца. Гемодинамика при ГКМП страдает преимущественно ввиду резкого снижения ударного объема как вследствие падения диастолического наполнения, так и вследствие обструкции тракта оттока (при довольно высокой фракции выброса).
- 3. Рестриктивная КМП (РКМП) — характеризуется нарушением диастолического расслабления (и как следствие — наполнения) ЛЖ и/или правого желудочка (ПЖ), может сопровождаться уменьшением их объема и долгое время сочетается с нормальной или почти нормальной систолической функцией. Гемодинамика при 194 РКМП страдает исключительно ввиду резкого снижения диастолического наполнения ЛЖ и, как следствие, сердечного выброса (при нормальной фракции выброса) [2].
По мере развития кардиологической теории и практики, благодаря открытию многих этиологических факторов и патогенетических путей развития КМП, появились и некоторые уточнения в понимании традиционных видов кардиомиопатий.
Так, собирательным образом были описаны ЭХО-кардиографические варианты ГКМП:
- традиционный (асимметричная и преимущественно диффузная гипертрофия межжелудочковой перегородки (МЖП) с обструкцией тракта оттока в систолу);
- европейский апикальный (гипертрофия верхушечных сегментов МЖП, без обструкции тракта оттока);
- азиатский апикальный (гипертрофия верхушечных сегментов МЖП и задней стенки ЛЖ (ЗСЛЖ), без обструкции тракта оттока);
- мидвентрикулярный (гипертрофия средних сегментов МЖП и ЗСЛЖ по типу «песочных часов», с обструкцией в средней части полости ЛЖ и перегрузкой верхушечной части полости ЛЖ);
- атипичный (асимметричная гипертрофия ЗСЛЖ, без обструкции тракта оттока в систолу);
- идиопатический гипертрофический субаортальный стеноз (отдельная, редкая форма ГКМП, при которой наблюдается асимметричная и преимущественно локальная (не диффузная, как при традиционном типе), только в субаортальном сегменте, гипертрофия межжелудочковой перегородки с обструкцией тракта оттока в систолу).
Для ГКМП, как ни для какой иной формы кардиомиопатий, очевидна генетическая подоплека заболевания. Генетические причины ГКМП описаны в многочисленных научных и практических работах. ГКМП как самостоятельное идиопатическое заболевание была описана в конце 50-х годов ХХ в. По данным Bonne et al. Первое сообщение о семейном характере заболевания относится к 1960 г. [3]. В дальнейшем установлено, что более 50% случаев ГКМП имеют семейный характер с аутосомнодоминантным типом наследования [4]. Картирование первого локуса (на хромосоме 14, где расположен ген тяжелой цепи бета-миозина (MYH7)), ответственного за развитие ГКМП, было осуществлено Jarcho et al. в 1989 г. [5]. Вскоре было установлено, что ГКМП является генетически гетерогенным заболеванием и к настоящему времени уже известно не менее 7 ядерных генов, мутации которых ассоциированы с ГКМП. Все эти гены кодируют белки, формирующие саркомер, поэтому ГКМП часто называют «заболеванием саркомера» [6]. Основными генетически изменяемыми при ГКМП белками являются: бета-миозин, сердечная изоформа тропонина Т, тропонин I, основная и регуляторная цепи бета-миозина, альфа-тропомиозин и миозин-связывающий белок С, альфа-сердечная изоформа актина [7].
Таким образом, ГКМП традиционно рассматривается как идиопатическое заболевание, при котором не должно быть иных гемодинамических условий развития диффузной или локальной гипертрофии миокарда (клапанного порока, артериальной гипертензии и др.).
Несколько сложнее сделать вывод о нозологической самостоятельности РКМП. Так, в связи с детальным описанием некоторых патогенетических типов развития рестрикции миокарда ЛЖ появились дополнительные рубрификации РКМП:
- РКМП, ассоциированная с фиброзом (наблюдается при выраженной ДДЛЖ у лиц пожилого и старческого возраста, а также при склеродермии);
- РКМП при инфильтративных заболеваниях миокарда (амилоидозе, метаболических синдромах, опухолях, прорастающих миокард и др.);
- РКМП при болезнях накопления (гемохроматозе, гликогенозах, болезни Фабри);
- РКМП при преимущественно эндомиокардиальных болезнях (тропическом эндомиокардиальном фиброзе, гиперэозинофильном синдроме, карциноиде, радиационных поражениях).
Несмотря на кажущуюся постоянной ассоциацию рестриктивных изменений сердца и каких-либо конкретных инфильтративных или фибротических/склер отических процессов при описанных состояниях для значительной части больных РКМП конкретные этиологические факторы не находятся и, в подавляющем большинстве клинических случаев, РКМП по-прежнему рассматривается как идиопатическое (генетически детерминированное) заболевание [8].
Благодаря прогрессивному развитию генетики были детализованы и выделены по этиопатогенезу некоторые подтипы ДКМП:
- семейная ДКМП с аутосомно-доминантным наследованием;
- семейная ДКМП с аутосомно-рецессивным наследованием;
- ДКМП в сочетании с мышечной дистрофией;
- ДКМП, сцепленная с полом.
ДКМП с аутосомно-доминантным наследованием. Наблюдается в 20–30% случаев всех ДКМП [9]. Возникает чаще на 3-м десятилетии жизни, манифестирует прогрессирующей сердечной недостаточностью (СН) и аритмиями. В семьях этих больных картированы 5 локусов с локализацией мутации в 9q13-q22, а также 1q32 и 10q21-q23 [10]. В последнем случае наблюдался также пролапс передней створки митрального клапана (ПСМК).
В более позднем возрасте у больных диагностируют семейную ДКМП с различными нарушениями проводимости, ассоциированную с патологией в хромосоме 1, а также с мутацией в 3p22-p25 [11], 2q31 и с патологией белков метавинкулина и адалина [12].
Выявляются также больные семейной ДКМП, у которых миопатия ассоциировалась с системными, иммунологическими расстройствами и миокардитом; в некоторых случаях ДКМП ассоциируется с нарушениями проводимости и мышечным дефектом перегородок сердца с выявлением патологического локуса в хромосоме 6q23 в области 3-сМ. Спектр мутаций гена ламина обнаруживали при ДКМП с нарушениями проводимости и мышечной дистрофией [13]. Возможность мутации гена актина различного типа находили как при ДКМП, так и при ГКМП [14].
ДКМП с аутономно-рециссивным наследованием также выявляется в ряде семей. Генетический локус при этом остается неидентифицированным, однако речь не идет о вовлечении митохондрий. При обследовании 118 добровольцев из 13 семей с ДКМП E. Arbustini et al. констатировали отсутствие особых фенотипических признаков и аутосомно-доминантный тип наследования в 11 семьях, а аутосомно-рециссивный — лишь в 2 семьях [15]. Имеется ряд экспериментальных моделей этой патологии на мышах, морских свинках, где показана возможность мутации гена саркогликана (трансмембранный гликопротеин, функционирующий в комплексе с дистрофином).
ДКМП в сочетании с мышечной дистрофией наблюдается нередко, развивается в результате мутации в гене дистрофина и состоит из следующих подтипов, известных преимущественно в неврологической практике:
- мышечная дистрофия Дюшена;
- мышечная дистрофия Беккера;
- особые неуточненные формы болезней.
ДКМП, сцепленная с полом, подразделяется на 2 типа:
1 тип — синдром Барта — развивается в детстве, проявляется нейтропенией, миопатией, замедленным ростом, ацидурией; больные умирают рано, нередко — от сепсиса;
2 тип — возникает в более зрелом возрасте (иногда у молодых мужчин) и быстро прогрессирует, обычно с повышением креатинфосфокиназы (КФК) и нетяжелой миопатией.
Таким образом, многие варианты ДКМП имеют определенный, в последние десятилетия генетически детерминированный генез. Тем не менее, далеко не всякая причина идиопатической дилатации сердца может быть легко типирована по приведенным выше генетическим локусам и отнесена к определенной группе.
Приведенные данные убедительно свидетельствуют, что для дилатационных кардиомиопатий не существует таких этиологических факторов, как воспаление сердечной мышцы, болезни перикарда, патологии клапанного аппарата сердца, следовательно, ДКМП, как и предполагается в классическом определении, являются идиопатической нозологией.
В последние годы в клиническую теорию и практику введено понятие «митохондриальной кардиомиопатии». Митохондриальная ДНК отличается от ядерной геномной ДНК тем, что она не имеет интронов (вставок), защитных гистонов и эффективной системы восстановления ДНК, частота мутаций в митохондриальной ДНК превосходит частоту таковых для ядерной ДНК в 10 раз. Присутствие различных количеств нормальной или мутировавшей митохондриальной ДНК в одних и тех же клетках и тканях объясняет потенциальную селективность вовлечения сердца, клиническую гетерогенность болезни у пациентов из одной семьи. Мутации митохондриальной ДНК находили также у больных ГКМП, причем с поздним развитием СН. Большинство ДКМП, связанных с мутациями митохондриальной ДНК, описаны лишь в отдельно взятой семье. Некоторые случаи митохондриальной ДКМП возникали при действии таких токсических агентов, как доксорубицин и зидовидин. Ультраструктурные и иммуноцитохимические исследования тканей, полученных при биопсии миокарда больных ДКМП, выявляли разнообразную патологию митохондрий: патологические включения, концентрические и тубулярные кисты, снижение антиферментной активности. При эндомиокардиальной биопсии у более чем 600 больных ДКМП с симптомами СН в 85 случаях обнаруживались изменения митохондрий, схожие с теми, что наблюдались при митохондриальных ДНК дефектах, протекавших с миопатиями.
Развитие ДКМП ассоциировано с некоторыми типичными клиническими проявлениями, объединяемыми в несколько так называемых митохондриальных синдромов:
- MELAS (митохондриальная миопатия, энцефалопатия, молочнокислый ацидоз и эпизоды, похожие на инсульт);
- MERRF (эпилепсия с миоклонусом и раздражение красных волокон);
- KSS — Kearns-Sayre Syndrome (прогрессирующая наружная офтальмоплегия, пигментная дегенерация сетчатки, с выпадением полей зрения; атаксия, мышечные парезы; дебильность; малый или карликовый рост, гипогонадизм; дилатация сердца, нарушения внутрижелудочковой проводимости по типу неполной или полной блокады правой ножки пучка Гиса, признаки двух- и трехпучковой блокады, переходящей в полную атриовентрикулярную блокаду);
- дефицит NAD-H коэнзим Q редуктазы (мышечная слабость, умственная отсталость, офтальмоплегия, лактатацидоз, при биопсии скелетной мышцы ragged-red волокна, накопление липидов и паракристаллов в митохондриях).
Описан целый ряд делеций и точечных мутаций митохондриальной ДНК, выделяют и наиболее чувствительные их участки, в результате изменений которых развивается как ДКМП, так и ГКМП. Делеции характерны для KSS, точечные мутации — при MELAS и MERRF A3243G и A8344G в tPHK. По данным T. Obayashiet al., возможно возникновение нескольких мутаций (до 8) у одного больного, при этом число их было закономерно выше, чем в контроле [16]. F. Muntoni et al. обращают внимание на роль мутаций митохондриальной ДНК в развитии СН, особенно у больных сахарным диабетом и низкорослых [17].
Из приведенных данных следует, что мутации митохондриальной ДНК могут сопровождать как ДКМП, так и ГКМП, а также быть единственной причиной дилатационного ремоделирования сердца вне определенных и сцепленных с наследованием кардиопатий. Следовательно, «митохондриальную кардиомиопатию» можно рассматривать и как самостоятельную нозологическую форму, развивающуюся вне известных причинных этиологических факторов сердечно-сосудистых заболеваний, и как отдельный этиологический фактор развития дилатации или гипертрофии сердца, и как следствие патологических процессов в миокарде. В случаях, когда достоверно установлено, что изменения митохондриальной ДНК предшествуют развитию ДКМП или ГКМП, можно говорить о первичной или истинной митохондриальной кардиомиопатии (МКМП), при этом клинической манифестацией МКМП будут дилатационные или гипертрофические изменения в сердце. При выявлении патологии митохондрий на фоне уже имеющейся болезни (например, сахарного диабета, хронической СН (ХСН), развившейся на фоне клапанного порока) правильно полагать, что митохондриальные изменения являются вторичными, развившимися вследствие патологических ультраструктурных изменений, вероятно нацеленных на репарацию.
Подводя итоги разделу, посвященному ДКМП, РКМП, ГКМП и МКМП, можно констатировать, что эти состояния могут рассматриваться как самостоятельные, идиопатические (преимущественно генетические) нозологические формы.
С другой стороны, показано, что при многих известных заболеваниях внутренних органов инфекционной, обменно-метаболической, токсической и другой природы происходит закономерное специфическое поражение миокарда с нарушением его функций, которое иногда приобретает некоторые черты описанных выше КМП. Такая неопределенность критериев современного рассмотрения и деления КМП привела к тому, что в 1995 г. экспертами ВОЗ и Международного общества и федерации кардиологов было рекомендовано использовать термин «кардиомиопатия» для всех случаев поражения миокарда, ассоциированных с нарушением его функции [18]. Согласно этой классификации, ВОЗ выделяет 6 групп КМП: 1) ДКМП; 2) ГКМП; 3) РКМП; 4) аритмогенная дисплазия ПЖ; 5) специфические КМП и 6) неклассифицируемые КМП (табл.)
Таблица 1. Классификация кардиомиопатий (ВОЗ, 1995)
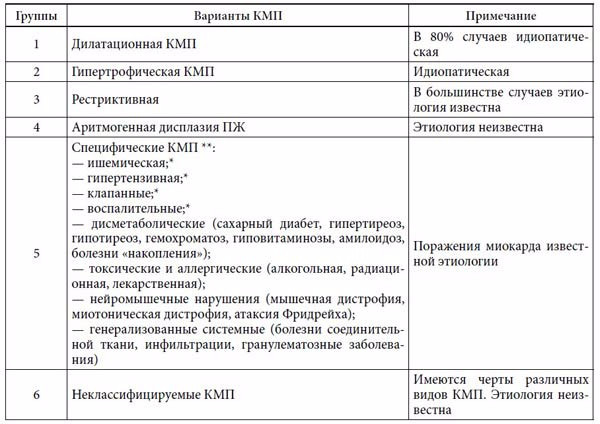
Приме ч а н и е: * — термины, которые должны быть заменены на традиционные: ишемическая болезнь сердца (ИБС), артериальная гипертензия (АГ), пороки сердца, миокардит; ** — «специфические (вторичные) поражения миокарда».
Приведенная классификация (ВОЗ, 1995) вызывала и вызывает серьезные критические замечания у большинства современных исследователей и врачей [19–21]. Во-первых, термин «кардиомиопатия» предлагается использовать для обозначения неограниченного количества заболеваний сердца известной и неизвестной этиологии, для которых характерно поражение миокарда, ассоциированное с нарушением его функции. Это приводит к неоправданно широкому употреблению понятия «кардиомиопатия» и полной утрате его нозологической самостоятельности. Во-вторых, вызывает большие сомнения правомочность включения в группу «специфических КМП» так называемых «ишемической», «гипертензивной», «клапанной» кардиомиопатий, которые, на самом деле, представляют собой лишь последствия известных заболеваний сердечно-сосудистой системы (ИБС, гипертоническая болезнь (ГБ), пороков сердца), осложненных сердечной недостаточностью и/или субъективно не манифестированной на определенном этапе дисфункцией желудочков. Правда, в рекомендациях экспертов ВОЗ подчеркивается, что к этим вариантам специфических КМП должны быть отнесены только те случаи заболевания, при которых выраженность нарушений функции сердца (например, дилатация полостей) не соответствует степени нарушений коронарного кровотока, клапанных поражений, уровню АД и т. п. Однако на практике решить вопрос о таком соответствии или несоответствии в большинстве случаев не представляется возможным [19, 22]. Учитывая сказанное, большинство авторов склоняются к тому, чтобы использовать термин «кардиомиопатия» только в тех случаях поражения миокарда, которые не являются следствием заболеваний клапанного аппарата (КА), перикарда, системной и легочной гипертензии, т. е. используя один из важнейших критериев J. Goodwin [21, 23, 24]. Поэтому не должны употребляться термины «ишемическая КМП», «гипертензивная КМП», «клапанная КМП». Вместо них целесообразно использовать традиционные термины: «ИБС», «пороки сердца», «артериальные гипертензии», «легочное сердце» и т. п. с указанием функционального класса хронической сердечной недостаточности. Мало того, все случаи невоспалительных поражений сердечной мышцы, этиология которых известна, должны обозначаться как «специфические (вторичные) поражения миокарда», а болезни сердечной мышцы воспалительной природы — как «миокардиты». Описанный подход деления вторичных поражений сердечной мышцы показали B. J. Maron [25] et al. в своей классификации от 2006 г. (рис. 1)
Приведенный подход B. J. Maron et al. также не лишен противоречий. Так, из всех классических КМП в генетической группе рассматривается лишь ГКМП, в то время как значительная часть ДКМП и РКМП по-прежнему может быть отнесена к наследственно-обусловленной, что убедительно продемонстрировано нами в выше изложенном материале.
Следует отметить, что ВОЗ при введении классификации в 1995 г. пользовалась инициативой научной группы P. Elliott, который в 2008 г. признал ошибочными некоторые положения классификации 1995 г. и предложил новое деление КМП, уже исключив из категории КМП такие нозологические формы, как ИБС, миокардиты, пороки и др. (рис. 2) [26]. Все КМП, по классификации P. Elliott, должны представлять один из нижеследующих типов структурного ремоделирования сердца: ДКМП, РКМП или ГКМП, в том числе ненаследуемые КМП.
Авторы статьи считают уместным по поводу основоположения классификации КМП преимущественно на «принципе ремоделирования» сделать следующее критическое замечание: клиническая практика показывает, что большинство ненаследуемых вторичных поражений миокарда при заболеваниях внутренних органов не сопровождаются дилатацией, констрикцией или рестрикцией вообще или длительное время. Если же такой процесс ремоделирования миокарда и развивается, то далеко не всегда он сопровождается отчетливыми признаками, позволяющими отнести ту или иную степень дилатации к дилатационной КМП, а ту или иную степень гипертрофии к гипертрофической КМП. Кроме того, группа приобретенных КМП, сцепленных с определенным заболеванием, в этой классификации по-прежнему представляет собой не что иное, как конкретную нозологию, в которой утрачивается самостоятельное нозологическое звучание КМП.
Именно в этом контексте уместно напомнить, что для ГКМП и ДКМП как первичных наследуемых заболеваний все же характерны определенные диагностические признаки:
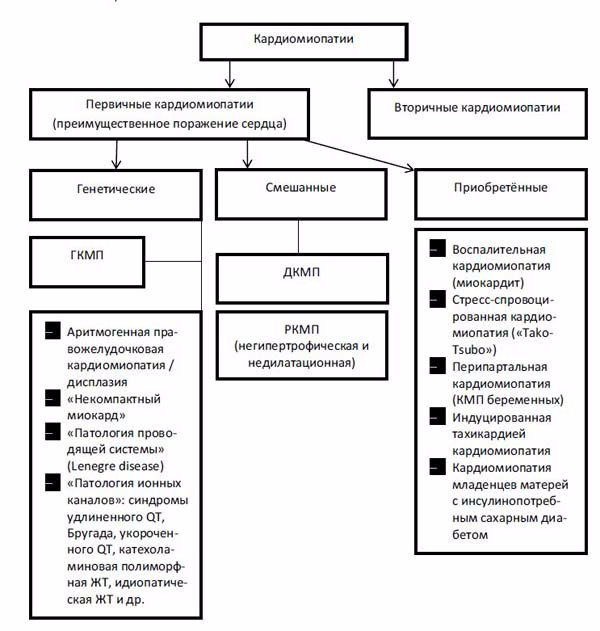
Рис. 1. Классификация кардиомиопатий B. J. Maron, J. A. Towbin et al., 2006 [25]
- для ГКМП — асимметричная ГЛЖ с толщиной МЖП от 15 мм и индексом асимметрии от 1,5; уменьшение размера полости ЛЖ менее 3,5 см; наличие митральной регургитации вследствие передне-систолического движения передней створки МК; так называемый митрально-септальный контакт (как систолический, так и диастолический);
- для ДКМП — равномерное истончение стенок ЛЖ (преимущественно менее 6–8 мм); идиопатическое расширение полости ЛЖ в диастолу более 60 мм (или >2,7 см/м2), снижение ФВ ЛЖ менее 45% и др.
И, наконец, несмотря на всеобъемлющий подход, приведенная классификация не описывает всего многообразия первичных и вторичных страданий сердечной мышцы, чему посвящены нижеприведенные разделы нашей статьи.
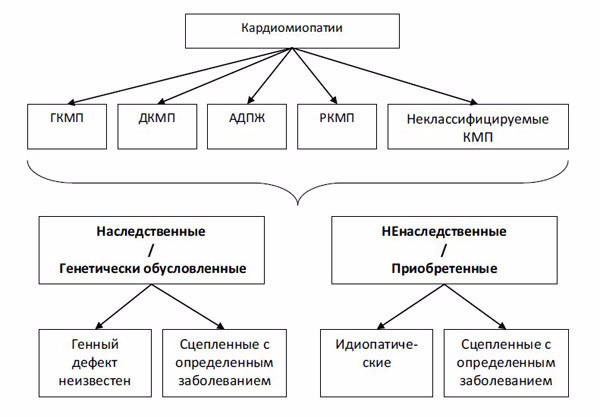
Рис. 2. Классификация кардиомиопатий (P. Elliott, 2008) [26]
На наш взгляд, в современных зарубежных классификациях утрачено одно важное положение — одним из ведущих факторов, определяющих структуру и функцию ЛЖ при генетически не детерминированных заболеваниях, является фактор метаболизма. Действительно, изменение обмена веществ при многих заболеваниях, не затрагивающих сердечно-сосудистую систему, приводит к трофическим нарушениям в миокарде и обусловливает изменение функционирования ЛЖ при первично интактном состоянии сердца. Не подлежит сомнению влияние нарушений обмена веществ в сердечной мышце и на диастолическое наполнение ЛЖ. Именно фактор метаболизма составляет основу для еще одной группы преимущественно вторичных поражений миокарда, сведения о которых мы приводим ниже.
Миокардиодистрофия (МКД) — группа вторичных поражений сердца, основой которых являются не связанные с воспалением, опухолью или первичной дегенерацией (отложением продуктов патологического синтеза) нарушения обмена веществ и дефицит энергии в миокарде, приводящие к обратимой на ранних стадиях развития дистрофии кардиомиоцитов и клеток проводящей системы сердца, что клинически проявляется различными расстройствами сердечной деятельности.
В номенклатуру болезней миокарда термин впервые был введен Г. Ф. Лангом (1936), но не в ограниченном морфологическом содержании понятия «дистрофия» (некробиоз, мутное набухание, жировое перерождение и др.), а как более широкое клиническое и патофизиологическое понятие, раскрывающее и подчеркивающее основополагающую роль процессов дистрофии на молекулярном уровне (патобиохимических, патобиофизических) в патогенезе функциональной недостаточности сердца, возникающей при ряде заболеваний, в том числе (и прежде всего) в случаях, когда морфологические изменения в миокарде не обнаруживаются или по выраженности и характеру не соответствуют выявленным функциональным нарушениям [27].
Учение Г. Ф. Ланга о миокардиодистрофии, особенно в варианте, вызванном переутомлением сердечной мышцы (дистрофия от гиперфункции), намного опередило время. Дальнейшие достижения медицинской науки позволили полностью подтвердить его правильность и конкретизировать механизмы формирования энергетического дефицита в миокарде на субклеточном и молекулярном уровнях [28]. Только недостаточным знакомством с этим учением можно объяснить тот факт, что болезни, относящиеся к группе МКД, в зарубежной медицинской литературе чаще обозначаются термином «миокардиопатия». Этот термин обдуманно и обоснованно был отвергнут Г. Ф. Лангом как «не дающий никакого представления о характере поражения миокарда». В современной отечественной кардиологии МКД рассматривается как вторичная (при различных заболеваниях), но относительно самостоятельная (по сущности патологического процесса) форма поражения миокарда, которую необходимо отличать от миокардита, опухолей сердца, болезней миокарда с неясными причинными факторами и патогенезом (так называемых кардиомиопатий) и первично-дегенеративных процессов в миокарде, связанных с патологическим отложением в нем различных продуктов патологического синтеза (при амилоидозе, гемохроматозе и др.). Выявляемые при перечисленных формах патологии дистрофические изменения миокарда, сопутствующие воспалению и склерозу, не рассматривают как самостоятельные и к группе МКД не относят [27, 29].
В основе развития МКД всегда лежит несоответствие между расходом энергии функционирующих структур миокарда, с одной стороны, и их восстановлением — с другой. Болезни и патологические состояния, являющиеся причинами возникновения такого несоответствия, при значительном их разнообразии могут быть систематизированы в три основные группы [30, 31].
Первая группа миокардиодистрофий включает болезни и патологические состояния, при которых развитие МКД связано с уменьшением поступления в организм и миокард веществ, необходимых для восстановления расходуемых структур в миокарде, либо кислорода, субстратов окисления или витаминов, которые обеспечивают процессы образования и утилизации энергии (дистрофия от первичного дефицита субстратов). Такова природа МКД при алиментарной дистрофии, гиповитаминозах (бери-бери), энтеритах с синдромом нарушенного кишечного всасывания, печеночной недостаточности (в связи с дефицитом белков), анемии, гипобарической гипоксемии (горной болезни) и в других случаях гипоксии миокарда (гипоксическая миокардиодистрофия), в том числе при легочной недостаточности. МКД вследствие ишемии миокарда при коронарной недостаточности (являющейся причиной недостатка и перерасхода метаболических и энергетических субстратов) рассматривается в соответствии с учением Г. Ф. Ланга в рамках ишемической болезни сердца. Примечательно, что Г. Ф. Ланг включал МКД как патофизиологическое понятие в нозологическую форму ИБС, подчеркивая вторичность дистрофии миокарда и первичность нарушений коронарного кровотока.
Вторую групп у миокардиодистрофий составляют болезни и патологические состояния, при которых нарушаются процессы клеточного дыхания, окислительного фосфорилирования и трансмембранного обмена катионов, в связи с чем снижаются образование энергии в миокарде и эффективность ее использования функционирующими структурами миокарда (дистрофия нарушенного потребления и/или усвоения) [32]. Такую природу имеют МКД при нарушениях электролитного баланса; при эндогенных (уремия) и экзогенных (цитотоксические яды, кардиотоксические лекарства, алкоголь, микробные токсины при острых и хронических инфекциях) интоксикациях. В эту группу отнесена МКД, развивающаяся вследствие нарушения регуляции процессов обмена веществ в миокарде при стрессе, поражениях головного мозга и периферических нервных структур (нейрогенная), дисфункции эндокринных желез (эндокринная), например, при сахарном диабете, тиреотокcикозе, патологическом климаксе, аддисонизме, гиперкортицизме [33].
Третья группа миокардиодистрофий объединяет патологические состояния, при которых несоответствие между расходом и восстановлением энергии функционирующих структур миокарда первично обусловлено значительным повышением энергозатрат в связи с избыточной нагрузкой на сердце (дистрофия повышенного потребления или дистрофия от гиперфункции). Существенную роль при этом может играть укорочение диастолы (в связи с тахикардией), в период которой главным образом и осуществляются восстановительные процессы [34]. В редких случаях МКД от гиперфункции развивается вследствие физического перенапряжения (например, при чрезвычайных спортивных нагрузках). По Г. Ф. Лангу, основными причинами этого одного из наиболее частых патогенетических вариантов МКД от перенапряжения являются артериальная гипертензия и пороки сердца; в состоянии гиперфункции могут быть участки сохраненного миокарда, замещающие функцию утраченной мышечной ткани при кардиосклерозе. По нашему мнению, в этом разделе уместно также отграничить первичные (условно не связанные с имеющейся болезнью, вызывающей перенапряжение миокарда) и вторичные (ассоциированные с ИБС, пороками, миокардитами, кардиосклерозом, гипертензией) МКД. Развивающаяся при перечисленных патологических состояниях МКД лежит в основе большинства случаев возникновения функциональной недостаточности сердца.
Приведенная систематизация причин МКД по основным патогенетическим механизмам в определенной мере условна, так как в каждом конкретном случае возникновения МКД эти механизмы нередко сочетаются [35].
Важно отметить, что факт клинической презентации МКД зависит от резервов дыхательной активности митохондрий, которые по мере прогрессирования патологии и/или гиперфункции постепенно снижаются: вначале при повышенном уровне их дыхательной функции в покое, а затем и при снижении этой функции, что совпадает с клиническими проявлениями сердечной недостаточности [27].
Классическое понимание термина «миокардиодистрофия» включает преимущественно вторичное нарушение обмена веществ в сердечной мышце. Описанную выше «митохондриальную патологию» необходимо дифференцировать на «митохондриальные кардиомиопатии» (установленные генетически детерминированные или приобретенные мутации митохондриальной ДНК) и «митохондриальную дисфункцию» (нарушение активности митохондрий вне установленных мутаций митохондриальной ДНК). Таким образом, мы предлагаем считать, что для кардиомиопатии более характерны генные дефекты, а для миокардиодистрофии — неопределенная на текущем этапе митохондриальная дисфункция.
Правомерность и обоснованность термина «миокардиодистрофия» подтверждается и рассмотрением патогенеза МКД на молекулярном уровне. Так, установлен тот факт, что структурная и функциональная перестройка миокарда как в процессе его гипертрофии, так и при формировании сердечной недостаточности детерминирована соотношением экспрессии определенных генов в хромосомах клеточного ядра, определяющих прежде всего трофические процессы. Показано, например, что снижение эффективности использования энергии гипертрофированным миокардом, обусловленное уменьшением общего количества и, следовательно, общей мощности Са-насоса в саркоплазматическом ретикулуме, происходит на фоне снижения содержания мРНК, кодирующей Са-АТФ-азу [36]. От экспрессии генов, кодирующих каждую из нескольких изоформ Na, К-АТФ-азы и миозина, зависит соотношение этих субстратов в кардиомиоцитах, которое и определяет эффективность функции миокарда [37].
Трофологические аспекты неразрывно связывают гормональные и генные механизмы регуляции. Так, механизмы формирования эндокринопатических МКД тесно ассоциированы с влиянием различных гормонов на экспрессию определенных генов. Так, трийодтиронин значительно повышает содержание в кардиомиоцитах мРНК, кодирующей синтез определенной изоформы Na-К-АТФ-азы, в то время как дексаметазон подавляет это его действие [38, 39]. Поскольку одной из важнейших составляющих нарушения обмена веществ в миокарде является нарушение гормонального баланса, мы считаем уместным уделить несколько более значительное внимание этому аспекту. Так, прямое или опосредованное гуморальное влияние на сердце оказывают практически все биологически активные вещества, содержащиеся в плазме крови. Согласно современным представлениям, регуляторная функция многих гормонов, вазоактивных пептидов (вазопрессин, предсердный натрийуретический фактор, адреналин, норадреналин, инсулиноподобный фактор роста, инсулин, кортизол, катехоламины, эндотелин, ангиотензин II, альдостерон), а также представительство и плотность рецепторных полей на мембранах кардиомиоцитов зависят прежде всего от структурно-функционального состояния клеточных регуляторных систем и от трофологических параметров [40–42]. Можно заключить, что при любом изменении клеточного метаболизма в миокарде, наиболее часто наблюдающемся при дистрофических процессах, нарушаются механизмы регуляции его деятельности [43].
Проведенный нами дополнительный анализ литературы позволил выделить в качестве наиболее частых (кроме диабета) и клинически значимых причин развития миокардиодистрофии такие состояния как: сахарный диабет [44], анемия [30, 45, 46], тиреотоксическое поражение миокарда [47–49], изменения сердца при физиологическом и искусственном климаксе [40, 50], инфекции и интоксикации.
В клинической практике и при проведении научных изысканий особое значение имеет проблема дифференцировки дистрофических процессов в миокарде от кардиосклеротических и ишемических. При таких состояниях, как хронические алкогольные интоксикации, тиреотоксикоз и постовариоэктомический синдром, не представлены первично склеротические и ишемические элементы патогенеза. В свою очередь, при наличии ишемии в миокарде имеют место трофические нарушения, которые на молекулярном уровне и уровне компартментов кардиомиоцита сходны с нарушениями на фоне тиреотоксикоза, дисовариального синдрома, алкогольных интоксикаций. В связи с чем для четкости понимания этиопатогенеза правильнее называть миокардиодистрофии, описанные Лангом и другими авторами, как вторичные поражения миокарда. Важность выделения вторичных поражений миокарда в отдельную группу основывается на известности этиологии и патогенеза, обратимости процессов в кардиомиоцитах, которые важны как для диагностики, так и для терапии состояний, вызвавших изменения в миокарде. Отдельной группой можно вынести первичные поражения миокарда неизвестной этиологии. ИБС, системные и легочные гипертензии, пороки сердца, миокардиты стоит также рассматривать отдельно, так как эти процессы сложно объединить по этиологии, патогенезу и подходами терапии этих заболеваний.
Учитывая изложенный материал, мы предлагаем решать терминологические проблемы КМП и МКД, возникающие в связи с медицинскими историческими и регионарными особенностями, нижеследующим образом.
1.«Кардиомиопатиями» (КМП) называть только первичные поражения миокарда, ассоциированные с нарушением функции сердца и не являющиеся следствием заболеваний коронарных артерий, клапанного аппарата, перикарда, системной и легочной гипертензии и воспалительного поражения сердечной мышцы (критерии J. Goodwin) [2].
2. Выделять следующие основные группы КМП:
- дилатационная кардиомиопатия (ДКМП);
- гипертрофическая кардиомиопатия (ГКМП);
- рестриктивная кардиомиопатия (РКМП);
- аритмогенная дисплазия ПЖ (АДПЖ);
- «некомпактный миокард» ЛЖ (НМЛЖ);
- «митохондриальная кардиомиопатия».
3. Предполагать, что термины «рестриктивная», «гипертрофическая», «дилатационная» КМП могут использоваться как этапные (на начальном этапе диагностического поиска), и в дальнейшем могут быть уточнены специфические причины (например, для РКМП — эндомиокардиальный фиброз, болезнь Леффлера и др. или для ГКМП — особые варианты вторичного ремоделирования миокарда, или для ДКМП — миокардит, кардиосклероз).
4. По мере уточнения природы идиопатических КМП необходимо «переносить» эти уточненные виды из раздела «кардиомиопатий» в раздел заболеваний с известной этиологией или в раздел «вторичных поражений сердечной мышцы». В случаях, когда невозможно выяснить причину характерных изменений сердца, рекомендовано сохранять термин «кардиомиопатия».
5. Считать целесообразным не вносить в понятие «кардиомиопатия» вторичные поражения миокарда, ассоциированные с врожденными или приобретенными нарушениями сердечного ритма (например, посттахикардитическая кардиопатия) и проводимости (например, болезнь Lenegre).
6. Поражения миокарда известной этиологии (при интоксикациях, гиповитаминозе, дисгормональных процессах и т. д.) обозначать как «специфические» (вторичные) поражения сердца или «кардиопатии» (не непосредственные и/или изолированные поражения миокарда, а поражение всего сердца) и не употреблять по отношению к ним термин «кардиомиопатии».
7. Устоявшиеся в клинической практике выражения «ишемическая», «тахикардическая», «токсическая», «дисгормональная» и т.п также целесообразно сопровождать термином «кардиопатия», исключая составляющую термина «мио» из определения для того, чтобы избежать совмещения с понятием «кардиомиопатия» (отдельная идиопатическая, проградиентная нозологическая форма).
8. К специфическим (вторичным) и тем более к первичным поражениям миокарда не следует относить случаи ИБС, системной и легочной гипертензии, пороки сердца и миокардиты, являющиеся самостоятельными нозологическими формами и вызывающими вторичное, но присущее конкретной нозологии страдание сердца/ миокарда.
9. Термин «миокардиодистрофия» рекомендовать использовать при любой нозологической форме в качестве отражения предположительно обратимых дистрофических патогенетических компонентов и акцентуации на дисметаболических процессах в сердечной мышце. Целесообразно употреблять термин «миокардиодистрофия» как этапный при любом вторичном поражении миокарда до момента улучшения трофических процессов в миокарде (подчеркивая обратимость дистрофического процесса). В случаях, когда на длительном промежутке времени не наблюдается восстановления трофических процессов в миокарде, такому патологическому состоянию сердечной мышцы можно присваивать категорию «кардиомиопатия», подчеркивая необратимость и проградиентность патологического процесса.
Автор статьи надеется, что изложенный материал представляет теоретический и практический интерес для многих кардиологов и будет признателен за обсуждение и критические отклики читателей как на страницах журнала «Кардиология», так и в материалах других изданий.
Литература
- Brigden W. Uncommon myocardial diseases / Th e noncoronary cardiomyopathies // Lancet. 1957. Vol. 2. P. 1179–1184.
- Goodwin J. F. Br. Heart J. 1982. July. Vol. 48(1). P. 1–18.
- Bonne G., Carrier L., Richard P. et al. Familial hypertrophic cardiomyopathy: from mutations to functional defects // Circ Res. 1998. Vol. 83. P. 580–593.
- Oakley C., Singh L., Brown L., Hambly B. Structural eff ects of hypertrophic cardiomyopathy mutations on sarcomeric proteins // Biophysical Journal. 2003. Vol. 84 (2). P. 452a.
- Jarcho J. A., McKenna W., Pare J. A. P. Mapping a gene for familial hypertrophic cardiomyopathy to chromosome 14ql // New Engl. J. Medicine. 1989. Vol. 321. P. 1372–1378.
- Th ierfelder L., Watkins H., MacRae C. et al. Alpha-tropomyosin and cardiac troponin T mutations cause familial hypertrophic cardiomyopathy: a disease of the sarcomere // Cell. 1994. Jun 3. Vol. 77(5). P. 701– 712.
- Mogensen J., Klausen I. C., Pedersen A. K. et al. Alpha-cardiac actin is a novel disease gene in familial hypertrophic cardiomyopathy // J. Clin. Invest. 1999. May 15. Vol. 103 (10). P. R39–43.
- Nihoyannopoulos P., Dawson D. Restrictive cardiomyopathies // Eur. J. Echocardiogr. 2009. Vol. 10 (8). P. iii23–iii33.
- Gregori D., Rocco C., di Lenarda C. et al. Estimating the frequency of familial dilated cardiomyopathy // Circulation. 1996. Vol. 94. P. 1–6.
- Bowles K., Gajarski R., Porter P. et al. Gene mapping of familial autosomal dominant DCMP to chromosome10q21–23 // J. Clin. Invest. 1996. Vol. 98. P. 1355–1360.
- Olson T., Keating M. Mapping a cardiomyopathy locus to chromosome 3p22-p25 // J. Clin. Invest. 1996. Vol. 97. P. 528–532.
- Maeda M., Holder E., Lowes B. et al. DCMP associated with defi ciency of the citosceletal protein metavinculin // Circulation. 1997. Vol. 95. P. 17–20.
- Bonne G., Muchir A. Spectrum of mutations in laminin A/C gene implicated in a new form of DCMP with conduction defects and muscular dystrophy // Circulation. 1999. Vol. 100 (18). P. 255.
- Olson T., Doan T. Hypertrophic and dilated CMP are caused by mutations in the cardiac actin gene // Circulation. 1999. Vol. 100 (18). P. 3256.
- Arbustini E., Diegoli M., Pilotto A. et al. Mitochondrial DNA mutations and CMP // Advances in cardiomyopathies / eds F. Camerini et al. Springer, 1997. P. 117–127.
- Obayashi T., Tsuji K., Tanaka A. et al. Mitochondrial DNA mutation as a cause of heart failure // Eur. J. Heart Failure. 1999. Vol. 1 (1). P. 72.
- Muntoni F., Wilson L., Marrosu M. et al. A mutatio in the dystrophin gene selectively aff ecting dystrophin expression in the heart // J. Clin. Invest. 1995. Vol. 96. P. 693–699.
- Richardson P., McKenna W., Bristow M. et al. Report of the 1995 WHO/ISFC Task Force on the Defi nition and Classifi cation of Cardiomyopathies // Circulation. 1996. Vol. 93. P. 841–842.
- Габрусенко С. А. Гипертрофическая кардиомиопатия: современное состояние проблемы // Болезни сердца и сосудов. 2006. № 1. С. 12–16.
- Горячева А. А., Хадарцев А. А. Особенности применения милдроната у больных миокардиодистрофией // Вестник новых медицинских технологий. 2007. Т. XIV, № 2. С. 201–201.
- Миронов С. А. Сравнительная оценка методов функциональной диагностики в выявлении различных типов диастолической дисфункции // Саратовский научно-медицинский журнал. 2009. Т. 5, № 2–2. С. 207–211.
- Годило-Годлевский В. А., Лубашев Я. А., Наговицын А. В. Клинико-экспертные подходы к диагностике некоронарогенных поражений миокарда // Военно-медицинский журнал. 2007. Т. 328, № 4. С. 53–56.
- Кушаковский М. С. Метаболические болезни сердца (Миокардии — миокардозы — миокардиодистрофии — кардиомиопатии). СПб.: Фолиант, 2000. 127 с.
- Ройтберг Г. Е., Струтынский А. В. Внутренние болезни. Сердечно-сосудистая система. М.: Бином, 2003. 856 с.
- Maron B. J., Towbin J. A., Th iene G. et al. Contemporary Defi nitions and Classifi cation of the Cardiomyopathies. An American Heart Association Scientifi c Statement From the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention // Circulation. 2006. Vol. 113. P. 1807–1816.
- Elliott P., Andersson B., Arbustini E. et al. Classifi cation of the cardiomyopathies: a position statement from the european society of cardiology working group on myocardial and periacardial diseases // Eur. Heart J. 2008. Vol. 29, N 2. P. 270–276.
- Коваленко В. Н., Несукай Е. Г. Некоронарогенные болезни сердца: практическое руководство / под ред. В. Н. Коваленко. Киев: Морион, 2001. 480 с.
- Хохлов А. Л., Лейнова Е. В. Изменения сердечно-сосудистых осложнений при тиреотоксикозе у пожилых пациентов на фоне медикаментозной терапии // Проблемы стандартизации в здравоохранении. 2008. № 1. С. 169.
- Окороков А. Н. Диагностика болезней внутренних органов. Т. 8: Болезни сердца и сосудов. М.:Медлит, 2003. 465 с.
- Гончарова Е. В., Говорин А. В., Кузьмин А. Г. и др. Структурно-функциональные показатели миокарда у больных хронической железодефицитной анемией // Кардиология. 2008. Т. 48, № 5. С. 46–50.
- Чепурная А. Н., Сафуанова Г. Ш., Никуличева В. И. и др. Гемическая кардиомиопатия у больных железодефицитной анемией // Кардиоваскулярная терапия и профилактика. 2008. Т. 7, № S22. С. 400–401.
- Тагильцева Н. В., Изможерова Н. В., Попов А. А. и др. Частота сердечно-сосудистых заболеваний у женщин с нарушениями углеводного обмена в климактерии // Эфферентная терапия. 2007. Т. 13, № 1. С. 102.
- Тармонова Л. Ю., Шутов A. M. Анемия и дисфункция почек у больных пожилого и старческого возраста с диастолической сердечной недостаточностью // Клиническая геронтология. 2007. Т. 13, № 11. С. 8–12.
- Dimopoulos K., Diller G. P., Giannakoulas G. et al. Anemia in adults with congenital heart disease relates to adverse outcome // J. Am. Coll. Cardiol. 2009. Vol. 22. P. 2093–2100.
- Adams K. F. Jr., Piña I. L., Ghali J. K. et al. Prospective evaluation of the association between hemoglobin concentration and quality of life in patients with heart failure // Am. Heart J. 2009. Vol. 6. P. 965–971.
- Daly M. J., Wilson C. M., Dolan S. J. et al. Reversible dilated cardiomyopathy associated with postpartum thyrotoxic storm // QJM. 2009. Vol. 3. P. 217–219.
- Шнейдер О. В., Обрезан А. Г., Макеева Е. Д. и др. Влияние структурных полиморфизмов генов ангиотензинпревращающего фермента, ангиотензиногена, эндотелиальной синтетазы оксида азота и рецептора брадикинина 2 типа на состояние миокарда у спортсменов и больных гипертонической болезнью // Цитология. 2004. №1. С. 69–78.
- Londhey V. A., Kamble U. S., Limaye C. S. et al. Irreversible dilated cardiomyopathy due to thyrotoxicosis // J. Assoc. Physicians India. 2006. Vol. 54. P. 575–576.
- Roffi M., Cattaneo F., Brandle M. Th yrotoxicosis and the cardiovascular system // Minerva Endocrinol. 2005. Vol. 2. P. 47–58.
- Mattar C. N., Harharah L., Su L. L. et al. Menopause, hormone therapy and cardiovascular and cerebrovascular disease // Ann Acad Med Singapore. 2008. Vol. 1. P. 54–62.
- Марченко Е. Н., Козиолова Н. А., Смирнова Е. Н. Новый алгоритм ведения больных тиреотоксической миокардиодистрофией, осложненной хронической сердечной недостаточностью // Кардиоваскулярная терапия и профилактика. 2009. № S2. С. 202a–202.
- Ngo A. S., Lung Tan D. C. // Th yrotoxic heart disease. Resuscitation. 2006. Vol. 2. P. 287–290.
- Олесова В. М., Маркатюк О. Ю., Юрова Ю. Ю., Обрезан А. Г. Метаболизм миокарда и препараты метаболического действия // Кардиология. 2013. № 1. С. 66–71.
- Бицадзе Р. М., Дорофейков В. В., Обрезан А. Г. Метаболические особенности сердечно-сосудистой патологии у больных сахарным диабетом 2 типа // Вестн. С.-Петерб. ун-та. Сер. 11. Медицина. 2009. Вып. 1. С. 3–10.
- Шевченко Ю. Л., Бобров Л. Л., Обрезан А. Г. Диастолическая функция левого желудочка сердца. М.: ГЭОТАР-МЕД, 2002. 240 с.
- Kubo T., Kitaoka H., Terauchi Y. et al. Hemolytic anemia in a patient with hypertrophic obstructive cardiomyopathy // J. Cardiol. 2010. Vol. 1. P. 125–129.
- Van de Donk N. W., America Y. G., Zelissen P. M., Hamer P. J. Takotsubo cardiomyopathy following radioiodine therapy for toxic multinodular goitre // Neth. J. Med. 2009. Vol. 10. P. 350–352.
- Карась А. С., Обрезан А. Г. Щитовидная железа и сердце // Клиническая и экспериментальная тиреоидология. М., 2009. № 3. С. 37–42.
- Карась А. С., Обрезан А. Г. Влияние гормонов щитовидной железы на сердце: молекулярные, клеточные, тканевые и органные аспекты (обзор литературы) // Вестн. С.-Петерб. ун-та. Сер. 11. Медицина. 2009. Вып. 4. С. 28–35.50.
- Ниаури Д. А., Обрезан А. Г., Ульянец М., Амелина А. А. Метаболический синдром у женщин в постменопаузе // Медицина XXI век. № 5 (6). 2007. С. 55–61.
Источник: Обрезан А.Г. Дискуссионные вопросы кардиологии: кардиомиопатия или миокардиодистрофия \\ Медицинский Алфавит: Кардиология-2 – 2014 - № 14 (203), C 12-20
