
Сахарный диабет (СД) является одним из самых распространенных эндокринных заболеваний. Согласно международным статистическим данным в настоящее время СД страдает 194 млн человек, а к 2025 г. этот показатель увеличится до 330 млн [1, 2]. При этом СД 2-го типа составляет 85–90 % от общего числа пациентов, страдающих СД. Пропорционально росту заболеваемости диабетом растет число его хронических осложнений. Ежегодно от осложнений СД умирает 5,5 % больных, уровень смертности среди них в 2–4 раза превышает таковой среди лиц без нарушений углеводного обмена, а продолжительность жизни у таких пациентов на 7–10 лет меньше, чем у лиц без диабета [1, 3].
Невозможно рассматривать СД как синдром гипергликемии; это заболевание, которое приводит к системному нарушению обмена веществ. Нарушение обмена веществ при СД происходит в основном из-за тканевой инсулиновой недостаточности. При этом расстраивается обмен не только углеводов, но и жиров, и белков, происходят изменения нейрогуморальных факторов и ионного обмена, что обусловливает целый ряд метаболических нарушений. Метаболические нарушения сопровождаются многочисленными функциональными расстройствами. Известно, что у больных сахарным диабетом имеется выраженная взаимосвязь между показателями углеводного и липидного обмена, гликозилированием белков, перекисным окислением липидов (ПОЛ), активностью антиоксидантной системы, системой гомеостаза. Особое значение имеет нарушение кислородотранспортной функции крови, свертывающей системы крови и как следствие — развитие микроангиопатий [4–11].
Инсулинорезистентность (ИР) — типичный признак у больных СД 2-го типа. ИР является первичным нарушением, приводящим к гипергликемии. Даже при выраженной резистентности тканей к инсулину нарушения регуляции углеводного обмена не происходит, пока не возникнут дефекты в компенсаторных механизмах повышения секреции инсулина. При ИР состояние нормогликемии поддерживается путем компенсаторного повышения секреции инсулина. Гиперинсулинемия (ГИ) нарастает по мере развития ИР. В настоящий момент не существует единых общепринятых критериев ГИ. Предлагается считать ГИ состояние, когда концентрация иммунореактивного инсулина в плазме крови натощак составляет 25,0 мкМЕ/мл, а также если его уровень через 2 ч после нагрузки глюкозой превышает 25,0–28,0 мкМЕ/мл [12].
Исследование иммунореактивного инсулина позволяет судить о секреции эндогенного инсулина только у больных, не получающих препаратов инсулина и не получавших их ранее, поскольку к экзогенному инсулину образуются антитела, искажающие результаты исследования. Пороговые уровни иммунореактивного инсулина крови, по данным различных авторов, находятся в пределах 11,0–28,0 мкМЕ/мл (чаще используют 11,0–15,3 мкМЕ/мл) [12, 13]. Со временем ресурсы В-клеток истощаются. Относительный дефицит инсулина при СД меняет биохимическое превращение глюкозы. При СД активизируется сорбитоловый путь метаболизма глюкозы. Скорость метаболизма глюкозы по сорбитоловому пути лимитируется активностью фермента альдозоредуктазы. В условиях гипергликемии и увеличения внутриклеточной концентрации глюкозы альдозоредуктаза активируется и из глюкозы образуется большое количество сорбитола. Сорбитол под влиянием полиолдегидрогеназы превращается во фруктозу:
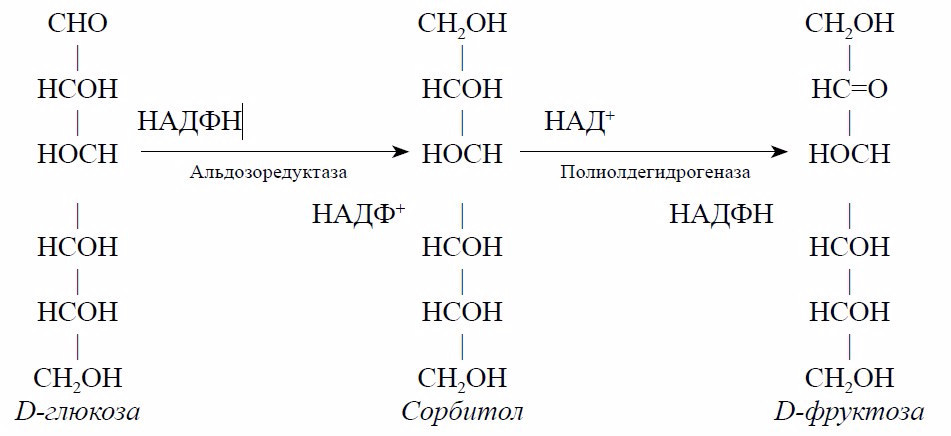
Сорбитол и фруктоза не проникают через клеточную мембрану. Их накопление в клетках создает гиперосмолярную среду, в результате чего вначале клетки набухают, а затем в них ингибируется Nа/К-АТФаза и клетки погибают [4, 14, 15]. Глюкоза используется в организме не только в качестве важнейшего источника энергии, но и для образования гликопротеинов.
В норме образование гликопротеинов контролируется специальными ферментами. При СД имеет место неферментативное гликозилирование белков, которое тем выраженнее, чем выше концентрация сахара в крови. Избыточное гликозилирование белков (гемоглобина, альбумина, коллагена, других белков базальной мембраны, белков хрусталика глаза, липопротеинов) сопровождается нарушением их функции. Уровень гликозилированных белков в крови служит надежным показателем качества регуляции метаболизма глюкозы. Активность гликозилирования белков и сорбитолового пути не регулируется гормонами и зависит только от уровня глюкозы крови. Конечные продукты гликозилирования (КПГ) вызывают поражение сосудистой стенки генерализованного характера. Образование КПГ на белках базальной мембраны сосудов приводит к ее утолщению, сужению просвета капилляров и нарушению их функции. Внеклеточное накопление КПГ изменяет структуру и функцию сосудов (снижение эластичности сосудистой стенки, изменение ответа на сосудорасширяющее действие оксида азота), способствует ускоренному развитию атеросклеротического процесса [4, 14]. При увеличении уровня HbA1c на 1 % риск развития сердечно-сосудистых заболеваний возрастает на 11 %.
В настоящее время постпрандиальную гиепргликемию рассматривают как самостоятельный независимый фактор риска развития сердечно-сосудистых осложнений при СД. Изолированная гипергликемия через 2 ч после стандартной углеводной нагрузки, превышающая 9 ммоль/л, при нормальной гликемии натощак (<6,1 ммоль/л) сопровождается двукратным увеличением риска сердечно-сосудистых осложнений и смерти [16]. При оценке риска сердечно-сосудистых осложнений у больных СД 2-го типа необходимо учитывать не только уровни гликемии натощак и HbA1с, но и величину постпрандиальной гипергликемии. С целью профилактики сердечно-сосудистых осложнений у больных СД рекомендовано достигать уровня гликемии плазмы крови ниже 6,0 ммоль/л натощак и 8,0 ммоль/л после еды, а гликозилированного гемоглобина — менее 6,5 % [17, 18].
Важным регулятором энергетического обмена являются гормоны гипофизарно-надпочечниковой системы и поджелудочной железы. При физиологическом соотношении гормональных взаимосвязей происходит торможение углеводного обмена с активацией основных ферментов гликонеогенеза и липидного обмена, что обеспечивает энергетическую стабильность организма. Инсулин стимулирует синтез липидов в жировых клетках, скелетных мышцах, печени; подавляет липолиз путем торможения гормонально-чувствительной липазы; регулирует высвобождение инсулина В-клетками поджелудочной железы. При СД отмечается усиление липолиза. В связи с относительной инсулиновой недостаточностью глюкоза не поступает в достаточном количестве в жировые клетки. Сам процесс липолиза приводит к ингибированию эффектов инсулина, происходит торможение тех процессов метаболизма, которые активирует инсулин, и клетка становится инсулинорезистентной [3, 5, 8]. Повышена секреция антагонистов инсулина, что также является важным фактором в развитии инсулинорезистентности и усугублении метаболических нарушений [5, 19, 20].
К антагонистам инсулина относят глюкокортикоиды, соматотропный гормон (СТГ), гормоны щитовидной железы, вызывающие гиперинсулинизм. Особое значение имеют глюкокортикоиды, которые повышают гликонеогенез, способствуют развитию гипергликемии, приводящей к ГИ, и уменьшают сродство рецепторов к инсулину. Эффекты контринсулярных гормонов не встречают адекватного противодействия со стороны инсулина. ГИ может быть следствием дисфункции гипоталамо-гипофизарной системы, а выраженная хроническая ГИ ее усугубляет [8, 10].
Сахарный диабет 2-го типа вследствие ГИ приводит к нарушению трансмембранных ионообменных механизмов. Отмечается увеличение концентрации Na+ и Са2+ внутри гладкомышечных клеток сосудов, что сопровождается повышением чувствительности клеток к прессорному действию норадреналина и ангиотензина; это способствует возрастанию общего сосудистого сопротивления и АД. Инсулин стимулирует Na+–К+-АТФазный насос, регулирующий внутриклеточный и внеклеточный баланс калия. При СД внутриклеточное содержание натрия возрастает, а калия — уменьшается, что провоцирует развитие электрической нестабильности миокарда. Как связующее звено между нарушениями углеводного, липидного обменов при СД 2-го типа обсуждается мембранопатия с нарушением метаболизма внутриклеточного ионизированного кальция [13, 21]. Инсулин регулирует активность Са2+-АТФазы плазматической мембраны клеток. Нарушение гомеостаза внутриклеточных ионов кальция, по мнению ряда исследователей, ответственно за возникновение нарушения секреции инсулина и формирование инсулинорезистентности [21, 22].ионообменных механизмов. Отмечается увеличение концентрации Na+ и Са2+ внутри гладкомышечных клеток сосудов, что сопровождается повышением чувствительности клеток к прессорному действию норадреналина и ангиотензина; это способствует возрастанию общего сосудистого сопротивления и АД. Инсулин стимулирует Na+–К+-АТФазный насос, регулирующий внутриклеточный и внеклеточный баланс калия. При СД внутриклеточное содержание натрия возрастает, а калия — уменьшается, что провоцирует развитие электрической нестабильности миокарда. Как связующее звено между нарушениями углеводного, липидного обменов при СД 2-го типа обсуждается мембранопатия с нарушением метаболизма внутриклеточного ионизированного кальция [13, 21]. Инсулин регулирует активность Са2+-АТФазы плазматической мембраны клеток. Нарушение гомеостаза внутриклеточных ионов кальция, по мнению ряда исследователей, ответственно за возникновение нарушения секреции инсулина и формирование инсулинорезистентности [21, 22].
Гиперинсулинемия, являясь компенсаторной реакцией, поддерживающей нормальный транспорт глюкозы в клетки в условиях ИР, одновременно приводит к серии метаболических нарушений, конечным результатом которых являются развитие и прогрессирование атеросклероза. В последние годы считается, что инсулинорезистентность служит независимым фактором риска развития атеросклероза [23, 24]. Однако точный механизм, посредством которого ИР ускоряет развитие атеросклероза, остается неясным. Существует мнение, что влияние ГИ и ИР на развитие атеросклероза в значительной степени связано с воздействием на процессы свертывания крови. Отмечаются отклонения в системе гомеостаза, характеризующиеся гиперкоагуляцией, что может способствовать внутрикоронарному тромбозу, увеличением агрегации тромбоцитов, снижением фибринолитической активности, повышением синтеза и активности ингибитора активатора тканевого плазминогена-1 (PAI-1) [25, 26].
Нарушение метаболических взаимосвязей при СД вызвано также перекисным окислением липидов. В организме нарушается естественный баланс между прооксидантными и антиоксидантными факторами в сторону ПОЛ. Перекисное окисление липидов способствует возникновению гемодинамических нарушений, прогрессированию макро и микроангиопатий, реализует стрессорные и гипоксические повреждения миокарда [8, 10, 11]. Окисленные формы липопротеинов низкой плотности оказывают выраженное проатерогенное действие.
Окислительный стресс в клетках сосудистой стенки сопровождается накоплением в атеросклеротической бляшке продуктов, которые могут образовываться только в результате взаимодействия свободных радикалов кислорода с белками и липидами [8, 27, 28]. Продукцию свободных радикалов кислорода стимулирует также ангиотензин II. На окислительный стресс в сосудистой стенке ангиотензин II действует следующим образом: 1) усиливает продукцию свободных радикалов макрофагами человека, эндотелиальными и гладкомышечными клетками сосудов; 2) увеличивает перекисное окисление липопротеинов низкой плотности (ЛПНП) и 3) стимулирует экспрессию цитокинов (фактор некроза опухоли, провоспалительные цитокины, тромбоцитарный фактор роста) [8, 27]. Свободные радикалы, окисляющие ЛПНП, продуцируются тканевыми макрофагами, входящими в состав атеросклеротической бляшки. Образование свободных радикалов кислорода под действием ангиотензина II происходит преимущественно в гладкомышечных клетках сосудов. Основным ферментом, катализирующим образование супероксид-аниона (О2-) в гладкомышечных клетках, является НАДФН-зависимая оксидаза плазматической мембраны, которая продуцирует более 90 % радикалов О2 -. Принципиальное отличие НАДФН-зависимой оксидазы гладкомышечных клеток от оксидаз нейтрофилов заключается в том, что ее активность может регулироваться в широких пределах и этот фермент способен постоянно продуцировать О2 -. Ангиотензин II — мощный активатор НАДФН-зависимой оксидазы, способный увеличивать продукцию О2 - в гладкомышечных клетках на 800 %. Повышение продукции супероксид-аниона и гидроксид-аниона (ОН-) под действием ангиотензина II сопровождается экспрессией большого количества специфических генов.
Внутриклеточный окислительный процесс в результате гиперпродукции НАДФН-зависимой оксидазы приводит к экспрессии свыше 200 генов, большинство из которых регулируют процессы клеточного деления, роста и дифференцировки [7, 27]. В реакцию с О2- может вступить медиатор эндотелийзависимой вазадилатации — NO, в результате которой образуется пероксинитрит (ONOO-), не обладающий сосудорасширяющими свойствами. Следовательно, в условиях окислительного стресса активность NO снижена. ПОЛ в первую очередь подвержены полиненасыщенные жирные кислоты (ЖК) фосфолипидов мембран [28]. Методы определения ПОЛ в настоящее время требуют значительной модификации. Не существует ни одного стандартного лабораторного теста на ПОЛ, адаптированного для автоматических биохимических анализаторов, разрешенных к использованию в Российской Федерации.
При СД отмечается множество коагулологических отклонений: повышение фибриногена, фактора фон Виллебранда, факторов VII, VIII, X, снижение антитромбина III, кроме того, наблюдается угнетение фибринолиза, связанное с повышением ингибитора активатора плазминогена 1 типа (РАI-1), также усилена адгезия и агрегация тромбоцитов, в качестве причины чего рассматривается дисфункция эндотелия [8, 25, 29]. Повышенное образование перекиси липидов подавляет синтез простациклина, увеличивает тромбогенный потенциал крови, возрастает агрегация и адгезия тромбоцитов, развиваются микротромбозы [8, 29].
Повышенный уровень фибриногена при СД связан с макро- и микрососудистыми осложнениями. Гипервязкость крови при СД, возможно, обусловлена повышением уровня глобулинов, С-реактивного белка (CRP) и некоторых компонентов комплемента. Рост концентрации СRP в сыворотке крови отражает активность воспаления, которое еще до развития инфаркта миокарда или инсульта связано с активностью атероматоза. Воспаление играет важную роль на разных этапах атерогенеза. Это относится к прикреплению и миграции лейкоцитов сквозь эндотелий, что сопровождается повреждением сосуда. Концентрация маркеров воспалительного процесса имеет независимое прогностическое значение. Это прежде всего относится к CRP, определенного ультрачувствительным методом (hs-method), который можно считать чувствительным показателем повышенного выброса цитокинов в ответ на воспаление. В связи с этим повышение концентрации hs-СRP рассматривают как признак атеросклероза. Величина базового уровня hs-СRP имеет важное практическое значение, так как непосредственно связана с риском развития тяжелых сердечно-сосудистых заболеваний и осложнений инфаркта миокарда и инсульта [13, 30]. При hs-CRP 1 мг/л риск развития сосудистых осложнений минимальный, 1,1–1,9 мг/л — низкий, 2,0–2,9 мг/л — умеренный, более 3,0 мг/л — высокий [30, 31].
Важным прогностическим фактором риска возникновения осложнений сердечно- сосудистых заболеваний у больных СД 2-го типа является дислипидемия. Сдвиги липидного обмена, сохраняющиеся у больных СД 2-го типа после коррекции уровня глюкозы в крови, настолько характерны, что получили название «диабетической дислипидемии». Эта липидная триада, представляющая собой специфический вариант атерогенной дислипопротеинемии, который способствует развитию атеросклероза, независимо от повышения уровня общего холестерина и общей фракции липопротеинов низкой плотности. Наиболее характерными и общими признаками дислипидемии у больных СД 2-го типа являются следующие [8, 9]:
- повышение уровня триглицеридов;
- повышение уровня ЛПНП;
- снижение уровня холестерина «антиатерогенной» фракции — ЛПВП.
Наличие дислипидемии у больных с СД в 2–4 раза увеличивает риск сердечно-сосудистой заболеваемости и летальности [4, 29]. Дислипидемия может иметь место на стадии преддиабета (метаболический синдром), а может возникнуть как следствие СД 2-го типа [20, 31, 33]. Инсулинорезистентность приводит к усилению липолиза и высвобождению большого количества свободных ЖК из жировой ткани, что в сочетании с повышенным содержанием глюкозы в крови дает дополнительное количество субстрата для синтеза триглицеридов в печени (который идет по глицерофосфатному пути). Соответственно синтезируется большое количество ЛПОНП, богатых триглицеридами. Также нарушен их катаболизм в результате снижения активности внепеченочной липопротеинлипазы. У больных СД 2-го типа отмечается уменьшение отношения липопротеиновой липазы к печеночной липазе за счет сниженной активности липопротеиновой липазы, что способствует повышению катаболизма ЛПВП [15, 29]. Эти изменения в кинетике метаболизма ЛПВП при СД 2-го типа сочетаются с повышением транспорта триглицеридов из ЛОНП, что опосредованно приводит к снижению содержания антиатерогенных ЛПВП.
Среди качественных изменений липопротеинов выделяют следующие:
- наличие малых плотных ЛПНП (обладающие наибольшей атерогенностью);
- неферментативное гликозилирование апопротеинов, входящих в состав основных классов липопротеинов.
Этот процесс прямо зависит от повышенного уровня глюкозы в крови и приводит к образованию модифицированных (гликозилированных) липопротеинов. Модифицированные ЛПНП быстрее и легче захватываются макрофагами с образованием пенистых клеток в атероме. В результате модифицирования ЛПВП (гликозилирование апобелков, перекисное окисление, увеличение содержания в них триглицеридов и числа малых плотных ЛПВП) укорачивается время их жизни, снижается концентрация, в итоге нарушается обратный транспорт холестерина [9, 25, 34, 35].
Дислипидемия прямо и опосредованно участвует в патогенезе ангиопатий при СД. Гипертриглицеридемия, увеличение процентного содержания малых плотных ЛПНП, снижение концентрации ЛПВП составляют липидную триаду, которая способствует развитию атеросклероза независимо от повышения уровня общего холестерина и общей фракции холестерина ЛПНП [32]. СД ускоряет развитие атеросклероза, который является морфологической основой ишемической болезни сердца (ИБС) и цереброваскулярных заболеваний [9, 19, 26, 32, 36, 37]. Липидная триада при близком к нормальному значению холестерина — характерная особенность диабетической дислипидемии. Присочетании СД с ожирением ускоряется ишемическое поражение миокарда на фоне коронарогенных и метаболических нарушений. Гиперлипидемия вызывает повышение содержания в миокарде свободных жирных кислот (СЖК) с кардиотоксическим эффектом [8].
У больных СД с абдоминальным ожирением резко расширяется спектр повреждающих факторов метаболизма. Инсулинорезистентность, дислипидемия и гормональные нарушения способствуют развитию диабетической ангиопатии, артериальной гипертонии (АГ) и ИБС. По данным исследования UKPDS (UK Prospective Diabetes Study), повышение уровня ЛНП на 1 ммоль/л вызывает увеличение риска развития ИБС на 57 %, повышение ЛВП на 0,1 ммоль/л приводит к снижению риска ИБС на 15 %, повышение систолического АД на каждые 10 мм рт. ст. обусловливает повышение риска на 15 %, а повышение концентрации HbA1c на 1 % сопровождается увеличением риска на 11 % [38]. Высокий уровень ЛНП является наиболее сильным предиктором развития ИБС при сахарном диабете 2-го типа. Своевременное выявление и коррекция диабетической дислипидемии является важным условием снижения количества тяжелых осложнений и увеличения продолжительности жизни больных диабетом.
На основании изложенных данных можно заключить, что основными метаболическими изменениями у больных СД 2-го типа, приводящими к развитию сердечно-сосудистых осложнений, являются: инсулинорезистентность и гиперинсулинемия, дислипидемия, перекисное окисление липидов, коагулологические отклонения.
Литература
- American Diabetes Association; National Heart, Lung and Blood Institute; Juvenile Diabetes Foundation International; National Institute of Diabetes and Kidney Disease; American Heart Association. Diabetes mellitus: a major risk factor for cardiovascular disease // Circulation. 1999. Vol. 100. Р. 1132–1133. 9
- Wild S., Roglic G., Green A. et al. Global prevalence of diabetes: estimates for 2000 and projections for 2030 // Diabet. Care. 2004. Vol. 27. P. 1047–1053.
- Business Briefi ng: Europan Endocrine review. 2006. A report by W. J. M. Wientiens. P. 14.
- Балаболкин М. И., Клебанова Е. М., Креминская В. М. Лечение сахарного диабета и его осложнений. М., 2005. С. 274, 304–414.
- Балаболкин М. И. Диабетология. М., 2000. С. 672.
- Дедов И. И., Александров А. А. Диабетическое сердце: Causa Magna // Сердце. 2004. № 1. С. 5–8.
- Мкртумян A. M. Кардиоваскулярные осложнения сахарного диабета 2 типа и особенности коррекции углеводного обмена // Там же. 2003. № 6. С. 266–272.
- Соколов Е. И. Диабетическое сердце. М., 2002. С. 64–65, 68, 330–354, 416.
- Мамедов М. Н. Особенности липидных нарушений у больных сахарным диабетом 2-го типа: в каких случаях следует применять статины? // Кардиология. 2006. № 3. С. 90–95.
- Благосклонная Я. В., Шляхто Е. В., Бабенко А. Ю. Эндокринология. СПб., 2004. С. 398.
- Бокарев И. Н., Великов Б. К., Шубина О. И. Сахарный диабет. М., 2006. С. 79–82, 94–95.
- Зимин Ю. В. Происхождение, диагностическая концепция и клиническое значение синдрома ИР или метаболического синдрома Х // Кардиология. 1998. № 6. С. 71–81.
- Ройтберг Г. Е. Метаболический синдром. М., 2007. С. 122–123.
- Долгов В. В., Селиванова А. В., Ройтман А. П. и др. Лабораторная диагностика нарушений обмена углеводов: Метаболический синдром, сахарный диабет. М., 2006. С. 4–5, 60–62.
- Frenais R., Nazih H., Ouguerram K. еt al. In vivo evedence for the role of lipoprotein lipase activity in the regulation of apolipoprotein AI metabolism: a kinetic study in control subjects and patients with type II diabetes mellitus // J. Clin. Endocrinol. Metab. 2001. Vol. 86. P. 1962–1967.
- DECODE studygroup on the European Diabetes Epidemiology Group: Glucose tolerance and mortality: comparision of WHO and American Diabetes Association Diagnostic criteria // Lancet. 1999. Vol. 35. P. 617–621.
- Global Guidelines for Type 2 Diabetes. International Diabetes Federation. 2005.
- American College of Endocrinology. American College of Endocrinology Consensus Statement on Guidelines for Glycemic Control // Endocr. Pract. 2002. Vol. 8. Suppl. 1. P. 1–82.
- Коненко И. В., Суркова Е. В., Анциферов М. Б. Метаболический синдром с позиции эндокринолога: что мы знаем и что уже можем сделать // Пробл. эндокринол. 1999. № 2. С. 36–41.
- Костин В. И., Карпов Р. С. Качество жизни пациентов с кардиологическим синдромом Х // Клинич. медицина. 2001. № 1. С. 25–27.
- Балаболкин М. И. Роль инсулинорезистентности в патогенезе сахарного диабета 2 типа // Терапевтич. архив. 2003. № 1. С. 72–77.
- Kahn B. B., Flier J. S. Obesity and insulin resistance // J. Clin. Invest. 2000. Vol. 106. P. 473–481.
- Balkau B., Eschwege E. Insulin resistance: an independent risk factor for cardiovascular disease? // Diabetes Obesity. Metab. 1999. Suppl. 1. P. S23–S31.
- Biegelsen E. S., Loscalzo J. Endotehelial function and aterosclerosis // Coron. Artery Dis. 1999. Vol. 10. № 4. P. 241–256.
- Панченко Е. П. Ишемическая болезнь сердца и сахарный диабет — коварный тандем // Сердце. 2004. Т. 3. № 1. С. 9–12.
- Гуревич М. А. Особенности патогенеза и лечения ишемической болезни сердца, сердечной недостаточности и артериальной гипертонии у больных сахарным диабетом // Клинич. медицина. 2005. № 1. С. 4–9.
- Шевченко О. П., Праскурничий Е. А., Шевченко А. О. Метаболический синдром. М., 2004. С. 142.
- Климов А. Н., Никульчева Н. Г. Обмен липидов и липопротеидов и его нарушения. СПб., 1999. С. 65–69, 299–306.
- Дедов И. И., Шестакова М. В. Сахарный диабет. М., 2003. С. 58–60, 285.
- Кишкун А. А. Руководство по лабораторным методам диагностики. М., 2007. С. 405.
- Ridker P. M., Rifai N., Pfeffer M. A. et al. Braunwald E. for the Cholesterol and Recurrent Events (CARE) Investigators: Infl ammation, pravaststin, and the risk of coronary events after myocardial infarction in patients with average cholesterol levels // Circulation. 1998. Vol. 98. P. 839–844.
- Аметов А. С., Сокарева Е. В. Статины в управлении сахарным диабетом 2 типа // Рос. мед. журн. 2006. Т. 14. № 26. С. 1901–1904.
- Беляков Н. А., Сеидова Г. Б., Чубриева С. Ю., Глухов Н. В. Метаболический синдром у женщин. СПб., 2005. С. 48–55.
- Яфасов К. М., Дубянская Н. В. Дислипидемия при сахарном диабете II типа: патогенез и лечение // Кардиология. 2001. № 9. С. 74–77.
- Зайчик А. Ш., Чурилов Л. П. Патохимия (эндокринно-метаболические нарушения). СПб., 2007. С. 332–333.
- Токмакова А. Ю., Староверова Д. Н. Современные методы ранней диагностики диабетической макроангиопатии // Пробл. эндокринол. 2005. Т. 51. № 3. С. 39–40.
- Чазова Т. Е., Катхурия Ю. Б. Сахарный диабет и сердечно-сосудистые заболевания: факторы риска, клинические особенности, диагностика // Мед. помощь. 2001. № 5. С. 28–32.
- Stratton I., Adler A., Neil H. еt al. Association of glycaemia with macrovascular and microvascular complications of type 2 diabetes (UKPDS 35): prospective observational study // Br. Med. J. 2000. Vol. 321.P. 405–412.
Источник: Бицадзе Р. М, Дорофейков В.В., Обрезан А.Г. Метаболические особенности сердечно-сосудистой патологии у больных сахарным диабетом 2 типа. // Вестник Санкт-Петербургского университета Серия 11 медицина. Выпуск 1 июнь 2009 г. Санкт-Петербург.- С. 3-10
