
Àльвеолярный протеиноз легких, или альвеолярный фосфолипопротеиноз, является редким заболеванием, характеризующимся накоплением в альвеолах и мелких дыхательных путях белковолипидного вещества с последующим нарушением газообмена в легких и развитием прогрессирующей дыхательной недостаточности. Впервые это заболевание было описано в 1958 году S. H. Rosen [1]. Встречается альвеолярный протеиноз (АП) главным образом у лиц молодого и среднего возраста (20–50 лет), чаще у мужчин. Описаны случаи заболевания в детском и пожилом возрасте.
В патогенезе заболевания большое значение придается дисфункции альвеолярных макрофагов (АМ), которым принадлежит основная роль в клиренсе и катаболизме сурфактанта. Эти процессы зависят от активности колониестимулирующего фактора гранулоцитов и макрофагов (GM-CSF). Известно, что ростовые факторы (GM-CSF и M-CSF) влияют на созревание, дифференцировку и активацию альвеолярных макрофагов. В бронхоальвеолярном лаваже (БАЛЖ) больных АП обнаружены нейтрализующие антитела против GM-CSF [2]. Это позволило автору предположить, что идиопатический АП у взрослых людей является аутоиммунным заболеванием, при котором формирующийся дефицит GM-CSF приводит к снижению функциональной активности АМ. Следствием этого является снижение клиренса сурфактанта, накопление в альвеолах белковолипидных масс, лишенных поверхностно активных свойств. При этом происходит компенсаторная гиперплазия альвеолоцитов II типа и еще большая гиперпродукция сурфактанта. Кроме того, в отсутствие GM-CSF происходит увеличение другого ростового фактора (M-CSF), который приводит к альтернативной активации макрофагов и повышению экспрессии матричных металлопротеиназ (ММР-2 и ММР-9). Последние участвуют в резорбции ткани и развитии фиброза [3].
Известно также, что в организме человека существует семейство ядерных гормональных рецепторов (peroxisome proliferator-activated receptors — PPAR), которым отводится центральная роль в регуляции гомеостаза липидов и глюкозы, дифференциации клеток, иммунного и воспалительного ответа. В легких PPAR экспрессируются альвеолярными макрофагами, эпителиальными, эндотелиальными и гладкомышечными клетками. Активация рецепторов клеток натуральными и фармакологическими лигандами может приводить к нарушению экспрессии и активации PPAR и к развитию альвеолярного протеиноза [4].
У новорожденных детей развитие АП связывают с дефицитом сурфактантного протеина В (SP-B), вызванным мутацией его кодирующего гена. Дефицит SP-B приводит к внутриальвеолярному накоплению неполноценного протеина С (SP-C), что ингибирует функцию альвеолярных макрофагов и способствует еще большему угнетению клиренса сурфактанта [5].
Кроме генетической предрасположенности к АП отмечается роль инфекционного фактора в развитии этого заболевания у детей, страдающих хроническими заболеваниями легких, при которых выраженное нейтрофильное воспаление сопровождается высокой степенью окисления протеинов. Наиболее чувствительными из них оказались сывороточный альбумин, сурфактантный протеин А и 1-антитрипсин. Поэтому изобилие высокореактивных радикалов кислорода при хронических инфекционных заболеваниях легких может также вести к нарушению выработки сурфактанта и развитию альвеолярного протеиноза (6).
Выделяют три формы альвеолярного протеиноза: врожденную, приобретенную (идиопатическую) и вторичную. Врожденная форма АП характеризуется острым началом заболевания сразу после рождения с развитием респираторного дистресс-синдрома и быстрым прогрессированием заболевания. При генетическом исследовании обнаруживаются мутации генов, кодирующих сурфактантные протеины В и С (SP-B и SP-C), субъединицы GM-CSF рецептора или АВС-А3.
Приобретенная (идиопатическая) форма АП характеризуется кашлем, одышкой и прогрессирующей дыхательной недостаточностью. Для нее характерно наличие аутоантител к GM-CSF в сыворотке крови и БАЛЖ.
Вторичный АП встречается редко и включает случаи развития заболевания у лиц с иммунодефицитными расстройствами в результате хронической инфекции, у лиц со злокачественными новообразованиями и гемопоэтическими расстройствами, а также при воздействии неорганической пыли (острый силикоз), токсических паров и др. [7].
Клиническая картина АП аналогична многим интерстициальным заболеваниям легких, поскольку основным симптомом является медленно прогрессирующая одышка, которая может сопровождаться сухим или со скудной мокротой кашлем, болями в груди, быстрой утомляемостью, похуданием и субфебрильной температурой. Кровохарканье наблюдается редко. При прогрессировании заболевания наблюдаются признаки дыхательной недостаточности: цианоз, одышка, формирование «пальцев Гиппократа». Ухудшение течения АП во многом обусловлено присоединением бактериальной или грибковой суперинфекции и постепенно ведет к развитию легочной гипертензии и формированию легочного сердца [8].
Ранняя диагностика АП затруднена в связи с отсутствием патогномоничных клинических симптомов этого заболевания. Объективные лабораторные данные и результаты функциональных исследований неспецифичны. Так, при физикальном обследовании больного АП может определяться укорочение перкуторного тона, в основном над нижними легочными полями. При аускультации выявляется ослабленное везикулярное дыхание, иногда нежная крепитация. При исследовании функции внешнего дыхания — рестриктивные нарушения вентиляционной способности легких, снижение диффузионной способности легких. Рентгенологическое исследование органов грудной клетки выявляет двусторонние мелкоочаговые затенения, имеющие тенденцию к слиянию, расположенные в основном в средних и нижних легочных полях. На компьютерной томограмме легких можно выявить мелкоочаговые затенения и синдром «матового стекла».
Верификация диагноза основана на исследовании лаважной жидкости, в которой определяется многократное увеличение содержания белка. При изучении материала биопсии легочной ткани в альвеолах и бронхиолах выявляется ШИК — положительное вещество. Окрашивание реактивом Шиффа (или PAS -реакция) позволяет выявить гликоген, гликолипиды, нейтральные мукопротеиды и гликопротеиды, сиаломукопротеиды. Более специфичной считается реакция с иммунопероксидазой. Она всегда положительна при исследовании биопсийного материала легочной паренхимы и лаважной жидкости у больных первичным АП.
Диагноз альвеолярного протеиноза, как правило, ставится больным поздно. Выявление диссеминированного процесса в легких на рентгенограммах является поводом для госпитализации в противотуберкулезный диспансер, где больным проводится противотуберкулезная терапия без какого-либо эффекта. Все это задерживает раннюю диагностику заболевания и проведение патогенетической терапии.
«Золотым стандартом» в терапии АП в настоящее время считается лечебный бронхоальвеолярный лаваж, проведение которого возможно лишь в специализированных пульмонологических центрах. В свете последних данных о выявлении антител к GM-CSF в настоящее время больным АП рекомендуется проведение плазмофереза [2, 9].
Приводим описание случая, который демонстрирует трудности диагностики АП.
Пациент П., 22 лет, студент, житель Петербурга, поступил в пульмонологическое отделение Центра интенсивной пульмонологии и торакальной хирургии ГМПБ №2 18. 08. 2005 г. с жалобами на одышку инспираторного характера.
Из анамнеза известно, что до октября 2004 года пациент чувствовал себя практически здоровым человеком. Во время учебы в институте вел активный образ жизни и находился в постоянном психоэмоциональном напряжении. На этом фоне в конце октября внезапно повысилась температура тела до 40о, наблюдались озноб, покашливание, головная боль, онемение левой руки, боли в суставах и мышцах конечностей, общая слабость, снижение аппетита и похудание. На фоне приема жаропонижающих средств состояние в течение пяти дней нормализовалось. Пациент приступил к учебе и в течение последующих пяти месяцев чувствовал себя удовлетворительно.
Через пять месяцев у него внезапно возник приступ инспираторной одышки, длящийся около 15 минут, не связанный с какой-либо физической нагрузкой. Через месяц одышка повторилась, но при этом приобрела постоянный характер, усиливаясь к вечеру. Данное состояние продолжалось в течение недели. При этом больной не обращался к врачу и не принимал лекарственных средств.
Резкое ухудшение самочувствия наступило через два месяца (13. 04. 2005 г.), когда на фоне эмоциональной нагрузки появились одышка смешанного характера, сухой приступообразный кашель при контакте с холодным воздухом, выраженная слабость, сухость во рту, отсутствие аппетита, головокружение и дизартрия в виде «тягучей речи». Данные симптомы усилились на следующий день на фоне физического напряжения. Больной был осмотрен терапевтом и после проведения флюорографического исследования органов грудной клетки с подозрением на диссеминированный туберкулез легких и туберкулезный менингит 15. 04. 2005 г. госпитализирован в противотуберкулезный диспансер. При поступлении в стационар состояние больного было расценено как удовлетворительное. При объективном осмотре отмечался умеренно положительный симптом Кернига с обеих сторон, со стороны органов дыхания — равномерное ослабление везикулярного дыхания. Изменений со стороны других органов и систем обнаружено не было.
При обследовании в стационаре убедительных данных в пользу туберкулезной этиологии заболевания получено не было, хотя отмечен положительный (21. 04. 2005 г.) ИФА с туберкулезным антигеном и сомнительная реакция Манту с 2ТЕ -11мм (6. 05. 2005 г).
Рентгенологическое исследование легких (19. 04. 2005 г.) (рис. 1) выявило двусторонние малоинтенсивные очаговые и инфильтративные тени в верхних долях обоих легких, изменения интерстициальной ткани: периацинусную и междольковую инфильтрацию и за счет этого — снижение пневматизации легочной ткани. Корни структурны, синусы свободны.

Рис 1. Рентгенограмма органов грудной клетки (19. 04. 05 г.).
Фибробронхоскопия (17. 06. 2005 г.): двусторонний, более выраженный справа, катаральный эндобронхит.
Посев мочи и мокроты на БК от 20. 04. 2005 г. — отрицательный.
Клинический анализ крови (18. 04. 2005 г.): эритроциты — 4,5 1012/л, гемоглобин — 142 г/л, лейкоциты — 12,8 109/л, палочкоядерных — 2%, сегментоядерных — 59%, лимфоцитов — 33%, моноцитов — 6%, СОЭ — 10 мм/час.
Исследование спинномозговой жидкости от 25. 04. 2005 г.: реакция Панди (+), белок — 0,23 г/л (норма 0,15–0,33 г/л), цитоз — 1 106/л (норма до 5 106/л), содержание глюкозы — 3,0 ммоль/л (норма 2–3 ммоль/л).
УЗИ органов брюшной полости 28. 06. 05 г.: патологических изменений не выявлено, лимфатические узлы не увеличены.
ЭКГ (20. 04. 05 г.): ритм синусовый, неполная блокада правой ножки пучка Гиса.
С учетом выявленных изменений был поставлен диагноз «диссеминированный туберкулез легких» и проведено лечение, включающее рифампицин, этамбутол, пиразинамид, тубазид, офлоксацин, канамицин, глюкокортикоиды и витамины.
На фоне проводившейся в течение 3,5 месяцев массивной противотуберкулезной и дезинтоксикационной терапии исчезли явления дизартрии, однако усилились симптомы дыхательной недостаточности, развилась тугоухость и периодически возникало онемение левой руки.
При повторном рентгенологическом исследовании (03. 08. 2005 г.) обнаружено нарастание интерстициальных изменений в легких (рис. 2).
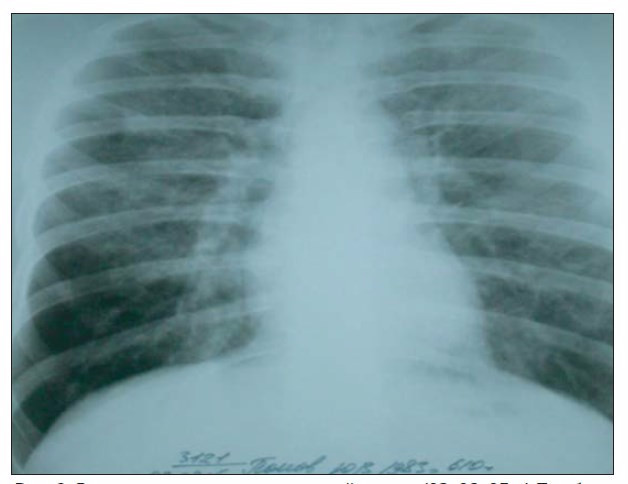
Рис 2. Рентгенограмма органов грудной клетки (03. 08. 05 г.).По обоим легочным полям — деформация легочного рисунка по типу интерстициального отека. Корни структурные, увеличенных лимфоузлов в корнях не определяется.
Результаты компьютерной томографии органов грудной клетки (10. 08. 2005 г.) показали картину диссеминированного процесса в легких без увеличения лимфатических узлов в корнях легких и средостении (рис. 3).

Рис 3. Компьютерная томография легких. Усиление и мелкоячеистая деформация легочного рисунка со снижением прозрачности легочных полей по типу «матового стекла».
Больной был выписан с диагнозом «Интерстициальный пневмофиброз неясной этиологии. Диссеминированный туберкулез легких, ВК(-)» и госпитализирован 16. 08. 2005 г. в Центр интенсивной пульмонологии и торакальной хирургии ГМПБ №2 для дальнейшего обследования и лечения. При поступлении пациент жаловался на одышку смешанного характера при умеренной физической нагрузке (подъем на второй этаж).
Детальное изучение анамнеза жизни больного позволило выявить, что его внутриутробное развитие протекало в неблагоприятных условиях. При рождении ребенок перенес родовую травму, а в возрасте 1,5 года двустороннюю пневмонию тяжелого течения, по поводу которой получал массивную антибактериальную терапию. В возрасте 12 лет больного беспокоили частые головные боли с рецидивирующими носовыми кровотечениями, при обследовании был поставлен диагноз «вегетососудистая дистония».
Из хронических интоксикаций пациент отмечает курение в течение последних шести лет по одной пачке сигарет в день. Наследственность не отягощена.
При объективном осмотре состояние было расценено как удовлетворительное. Обращали на себя внимание коробочный оттенок перкуторного тона над нижними отделами легких и ослабленное везикулярное дыхание над всеми легочными полями. Со стороны других органов и систем отклонений от нормы не отмечено.
Обследование выявило снижение напряжения кислорода в артериальной крови (РаО2 58,5 мм рт. ст.). Клинический и биохимический анализы крови соответствовали норме. Бодиплетизмография (18. 08. 2005 г.) показала отсутствие нарушений бронхиальной проходимости (ОФВ1 88,7% Д, ОЕЛ — 93,1 % Д, ЖЕЛ 85,6% Д , ООЛ 112% Д). Вместе с тем отмечалось умеренное снижение диффузионной способности легких: TLCO SB (mmol/min/kPa 6,88 (62% D), TLCO/VA (mmol/min/kPa/1 1,27 (57,6% D).
Таким образом, данные обследования свидетельствовали в пользу диссеминированного поражения легких. С целью более точной диагностики заболевания была произведена видеоторакоскопия с биопсией легкого и лимфатических узлов средостения, при которой получена картина альвеолярного протеиноза: альвеолы заполнены плотным мелкозернистым PAS-положительным веществом. Отмечены явления эмфиземы легких. Лимфатические узлы без изменений. На основании полученных данных был поставлен диагноз «Альвеолярный протеиноз легких. Острое течение. Дыхательная недостаточность II степени».
Данное заболевание явилось показанием к проведению тотального лаважа правого и левого легких с введением 5% раствора ацетилцистеина. После данной процедуры самочувствие больного значительно улучшилось, исчезла одышка. Показатели спирометрии (14. 12. 2005 г.) в пределах нормы: ЖЕЛ — 100,2% Д, ОФВ1 — 96,8% Д, ПОСвыд — 107,0% Д, МОС25 — 109,5% Д, МОС50 — 88,6% Д, МОС75 — 84,9% Д. Больной выписан из стационара в удовлетворительном состоянии.
Таким образом, представленный случай демонстрирует нетипичное течение альвеолярного протеиноза у молодого человека, поскольку диссеминированный процесс в легких сопровождался развитием менингеального синдрома, что при наличии положительного результата ИФА с туберкулезным антигеном и пробы Манту привело к ошибочной постановке первичного диагноза: «диссеминированный туберкулез легких, туберкулезный менингит». Длительная противотуберкулезная терапия не имела положительного эффекта и значительно отсрочила своевременную диагностику альвеолярного протеиноза, оказавшуюся возможной только в специализированном центре, где верифицирована данная патология и проведена адекватная терапия.
Детальный анализ настоящего заболевания показывает, что первые неврологические симптомы появились у пациента за полгода до клинических проявлений диссеминированного поражения легких и сопровождались развитием интоксикационного и суставного синдромов. Можно предположить, что причиной этому послужила инфекция (хламидийная, герпетическая и др.), развившаяся на фоне иммунологической недостаточности у пациента с длительным эмоциональным напряжением в анамнезе. Известно, что хроническая персистирующая инфекция (например, вызванная С. pneumonia) ведет к поражению эндотелия сосудов головного мозга, сердца и других жизненно важных органов. Характер течения инфекционного процесса определяется внутриклеточным паразитированием микроба в эпителиальных клетках и альвеолярных макрофагах в персистентной или цитоцидной формах [10]. Персистенция возбудителей в организме может приводить к формированию аутоиммунного процесса. Можно предположить, что в данном случае имела место персистенция возбудителей в альвеолярных макрофагах и эндотелии сосудов мозга, что способствовало нарушению кровообращения и появлению транзиторных неврологических расстройств. Дисфункция альвеолярных макрофагов могла способствовать в дальнейшем развитию АП. В то же время длительная персистенция внутриклеточного возбудителя способна послужить причиной образования аутоантител к гранулоцитарно-макрофагальному колониестимулирующему фактору и, следовательно, альвеолярног протеиноза.
Для решения вопроса о форме АП — вторичной, в результате хронической инфекции или идиопатической, связанной с наличием аутоантител к GM-CSF — пациенту рекомендовано провести иммунологическое исследование для выявления персистенции возбудителя (C. pneumoniae, C. trachomatis, Herpes simplex) и выявления антител к GM-CSF. Результаты исследований позволят определить дальнейшую тактику лечения больного. Так, при выявлении персистенции инфекционного возбудителя возможно применение иммуномодуляторов, а в случае подтверждения аутоиммунного процесса — плазмофереза. Базисной терапией альвеолярного протеиноза в настоящее время остается проведение лечебного бронхоальвеолярного лаважа с целью удаления накапливающегося в альвеолах белково-липоидного вещества.
Литература:
- Rosen S. H., Castleman B., Liebow A. A. Pulmonary alveolar proteinosis // N.Engl.J.Med, 1998, V.258, р.1123–1142.
- MazzoneP.J., Jane Thomassen M., Kavuru M.S. Pulmonary Alveolar Proteinosis: resent advances // Semin.Respir.Crit. Care Med. 2002, v. 23, N2, р. 115–126.
- Bonfield T.L., Swaisgood C.M., Barna B.P., Farver C.F., Kavuru M.S., Thomassen M.J. Elevated gelatinase activity in pulmonary alveolar proteinosis: role of macrophage-colony stimulating factor // J. Leukoc. Biol, 2006, V.79, N1, р. 133–139.
- Denning G. M., Stoll L. L., Peroxisome Proliferator — Actovated Receptors: Potential Therapeutic Targets in LUNG Disease // Pediatr. Pulmonol, 2006, v.41. №1. р.23–24.
- Kattan A. K., Bulagannawar P. S., Malik I. H. Congenital alveolar proteinisis // Saudi Med.J, 2004, v.25, N.10, р.1474–1477.
- Starosta V., Griese M., Protein Oxidation by Chronic Pulmonary Disease in Children // Pediatr.Pulmonol, 2006, V.41, N 1, р.67–73.
- Paschen Ch., Reiter K., Stanzel F., Teschler H., Griese M. Therapeutic lung lavages in children and adults // Respir.Research, 2005, V.3, N.138, р.1186–1465.
- Venkateshiah S. B., Thomassen M. G., Kavuru S. Pumonary alveolar proteinisis.Clinical manifestations and optimal treatment strategies // Treat. Respir. Med, 2004, V.3, N.4, р.217–227.
- Интерстициальные заболевания легких // Руководство для врачей. Под ред. М. М. Ильковича и А. Н. Кокосова. Нордмед-Издат, Санкт-Петербург, 2005, с. 560.
- Лобзин Ю. В., Ляшенко Ю. И., Позняк А. Л. Хламидийные инфекции (Руководство для врачей) // СПб., Фолиант, 2003, с.396.
Источник: А. Г. Обрезан, Е. В. Евсюкова, П. К. Яблонский, Т. А. Степаненко, О. А. Гергес «Альвеолярный фосфолипопротеиноз легких трудности дигностики описание клинического случая»/ Медицина XXI век, № 2 {3} 2006, с. 83- 86
